Abstract
Multiple myeloma is a plasma-cell dyscrasia presenting with generalized neoplastic changes in bones, accompanied by impaired haematopoiesis, susceptibility to infections, and end-organ damage. However, the clinical picture of myeloma might be quite different from the classic manifestation, patients can present with renal disease, dominating or precluding multiple myeloma features. Diagnosis and differential diagnosis demand renal tissue pathology evaluation, most often kidney pathology demonstrate cast-nephropathy, AL amyloidosis, and light-chain deposition disease. Light-chain proximal tubulopathy is less frequently reported variant of paraproteinemic kidney damage in patients mostly not demonstrating overt clinical features of multiple myeloma. This lesion is almost always induced by the excess of kappa light chains, excreted through the kidney and reabsorbed in the proximal tubule cells, which manifests with moderate CKD, and, rarely, with acquired adult Fanconi syndrome. We present two cases of myeloma with light-chain proximal tubulopathy. None of our two patients met multiple myeloma criteria at admission, and only kidney biopsy, which discovered light-chain proximal tubulopathy, prompted further work-up, in turn resulted in the diagnosis of multiple myeloma. Our cases demonstrate some peculiarities, making them rare even in the setting of light-chain proximal tubulopathy, which is rare itself. Case one is an extremely infrequent example of light chain lambda deposition in the proximal tubules, case two demonstrates acute kidney injury on the top of chronic kidney disease, unusual for the light-chain proximal tubulopathy. We conclude that kidney biopsy should be performed, unless contraindicated, in all patients with proteinuria and/or impaired kidney function of unknown origin; and that the chemotherapy with bortezomib-based regimens is effective for treatment of myeloma with light-chain proximal tubulopathy.
Key words
plasma cell dyscrasia, monoclonal proteins, kidneys, Fanconi syndrome
Introduction
Multiple myeloma (MM) is a plasma-cell dyscrasia presenting with generalized neoplastic changes in bones, accompanied by impaired haematopoiesis and susceptibility to infections. To distinguish MM from other plasma cell dyscrasias, the diagnosis is based on histologic, serologic, and radiographic features: bone marrow clonal plasma cells; monoclonal protein in the serum or urine; and end-organ damage, evidenced by renal impairment, hypercalcemia, anemia, or lytic bone lesions. Sometimes, however, the clinical picture of MM is quite different from the classic manifestation, which can cause diagnostic difficulties, thereby delaying treatment. Importantly, symptoms of kidney damage may dominate over MM symptoms, and even preclude overt MM, and the diagnosis of MM often results from the workup of unexplained renal disease. Renal disease clinical presentation in MM includes acute kidney injury (AKI), nephrotic syndrome, proteinuria and/or haematuria, arterial hypertension and chronic kidney disease (CKD), occasionally, patients with MM present with renal tubular dysfunction [1-8].
Renal involvement is mainly caused by the deposition of monoclonal immunoglobulin’s or fragments thereof, mostly light chains (LC), as organized (crystals, fibrils, microtubules) and non-organized deposits, involving all compartments of the renal parenchyma: glomeruli, tubules, interstitial space and vessels. Organized deposits induce cast-nephropathy, light-chain proximal tubulopathy (LCPT), AL amyloidosis, glomerulonephritis with organized microtubular monoclonal deposits, and cryoglobulinemic glomerulonephritis, while non-organized deposits lead to the light chain deposition disease or proliferative glomerulonephritis with monoclonal deposits of IgG/IgA. It appears that amino acid sequence of the monoclonal LC and other monoclonal proteins, defining inherent biochemical properties, is the primary determinant of the specific pattern of renal parenchymal deposition and clinical disease. Differential diagnostics is the major challenge, demanding pathology evaluation of kidney tissue, as the above mentioned lesions cannot be differentiated solely on the base of clinical features. Most often kidney pathology in patients with MM demonstrate cast-nephropathy, AL amyloidosis, and light-chain deposition disease, less frequently cryoglobulinemic glomerulonephritis proliferative glomerulonephritis can be seen [9-18].
LCPT is a unique entity which is less frequently reported as case reports and small case series. In LCPT the excessive LC, almost always of kappa type, are excreted through the kidney and reabsorbed in the proximal tubule cells, leading to tubular damage, sometimes resulting in the defects in acidification and concentration and, rarely, the features of complete acquired Fanconi syndrome (FS). Of interest, in many of the LCPT cases renal manifestations dominated in the clinical presentation, patients did not have typical features of MM, and the diagnosis was predicated upon thorough immunopathology assessment of renal biopsies material. Most of these patients present with moderate CKD, slowly progressing towards ESRD and rarely – to symptomatic myeloma, and according to the International Kidney and Monoclonal Gammopathy Research Group (IKMGRG) definition regarded as monoclonal gammopathy of renal significance (MGRS) cases [19-31].
As only very few series of LCPT have been published, the evidence for the treatment efficacy is very weak, and the results of the so-called novel anti-myeloma agents has not been evaluated in these patients. According to the recommendations of the IKMGRG treatment choices should be adapted to the degree of renal failure. Thus in patients with stages 1 to 3 CKD, chemotherapy should be considered to try to slow progression to ESRD, cyclophosphamide-, bortezomib-, or thalidomide-based regimens are the best options [31]. Treatment of LCPT patients, presenting with AKI is not described in the literature, however the study of LC-induced AKI in the patients with cast-nephropathy demonstrated substantial improvement of renal function, mainly in cases with less severe renal impairment [32].
Here we present two cases of MM, manifested with kidney disease, and diagnosed on the basis of immunopathology evaluation of renal tissue.
Case 1
Caucasian male, 61 y.o., admitted to our unit January 18 2016.
Main complains: general weakness, dizziness, back pain.
Previous medical history: inguinal hernia, prostate benign hyperplasia, mild arterial hypertension, myocardial infarction in 2005, benign colon neoplasm with laparoscopic sigmoid resection in 2010.
History of present illness: February 2015 during outpatient annual work-up proteinuria 1.8 g/L was found. December 2015 proteinuria increased to 5 g/L and he was referred to our clinic.
At admission: conscious, alert, oriented. Body temperature 36.7°C, RR 17 per minute, pulse regular 78 per minute, BP 115/70 mm Hg. Slightly obese (BMI 29). Skin clear, normally colored, no edema. Eyes clear. HEENT and neck otherwise normal. Peripheral lymph nodes not palpable. Joints: no swelling, movements not restricted. Vertebral and flat bones percussion non painful. Lungs: clear. Heart: regular rate and rhythm, no murmur. Abdomen soft, non-tender, bowel sounds normal. Liver, spleen and kidneys not felt. Urine output 1500 ml/24 hours.
Work-up: total blood count, blood chemistry, serum osmolality and pH, serum electrophoresis, serum immunoglobulin’s and coagulation tests - within normal range, infectious screening negative. Urine specific gravity 1017-1030, urine pH 5.5, proteinuria 5.8 g/24 hours, glycosuria not detected, urinary sediment unremarkable. Electrocardiogram - post-infarction cicatricle changes. Plain chest X-ray otherwise normal. Kidneys and abdomen ultrasound – nothing but mild hepatomegaly.
Kidney biopsy
Light microscopy: sections of formalin fixed paraffin-embedded tissue were stained with H&E, Masson’s trichrome, periodic acid-Shiff and Congo red. 43 glomeruli, 4 out of them totally sclerosed. The rest appear otherwise normal, without proliferative changes, capillary walls thin, single-contour (Figure 1). Mild interstitial fibrosis and tubular atrophy, up to 10% of parenchyma, diffuse-focal interstitial infiltration with inflammatory cells in the areas of interstitial fibrosis. Tubular epithelial dystrophy (Figure 2) without any casts in the tubular lumen. Arteries and arterioles otherwise normal. Congo red staining was negative.
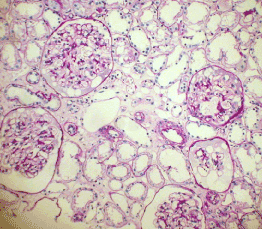
Figure 1. Case 1 - light microscopy. Otherwise normal glomeruli. PASx100
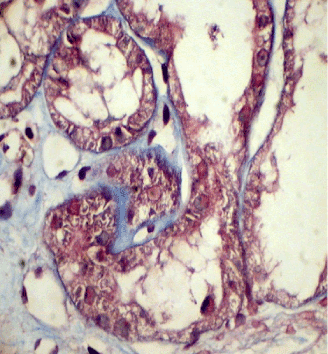
Figure 2. Case 1 - light microscopy. Tubular epithelial dystrophy. Masson x400
Immunofluorescence on unfixed cryo-sections with fluorescein conjugated anti IgA, IgG, IgM, C1q, C3, fibrinogen, lambda and kappa light chains antibodies. Positive (++) staining for lambda light chains in the resorptive granules within tubular epithelial cells (Figure 3), and negative staining for IgG, IgA, IgM, С3, С1q, fibrinogen and kappa light chains.
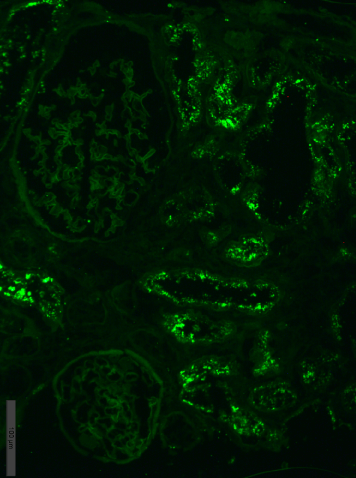
Figure 3. Case 1 - immunofluorescence. Lambda light chains fine granular expression
within tubular epithelial cells; x200
Pathologists conclusion: Focal global glomerulosclerosis 9%, immunofluorescence data suggestive for light-chain tubulopathy
Additional work-up
Skeletal X-ray: lytic lesions not detected.
Serum and urine immunochemistry: Bence-Jones protein lambda (traces in the serum with urinary excretion 5.96 g/24 hours). IgM deficiency. Cryoglobulines not found, CRP and β2-microglobuline within normal range.
Bone marrow smear: 63.5% of plasma cells, including 2- and 3-nuclear forms and cells with flammeous cytoplasm.
Final diagnosis: Multiple myeloma Bence-Jones lambda, light-chain tubulopathy with focal global glomerulosclerosis (CKD stage 2), secondary immunodeficiency
Treatment and follow-up: patient was referred to the hematology unit for chemotherapy and received 6 courses of bortezomib-based chemotherapy. At the latest follow-up visit in August 2016 he was doing well, his Bence-Jones lambda protein excretion decreased to 0.9 g/24 hours, serum ctreatinine remained normal.
Case 2
Caucasian female, 57 y.o., admitted to our unit April 1 2016
Main complains: general weakness, fatigue.
Previous medical history: tonsillectomy; tuberculosis as a child, received specific therapy and never recurred; cystitis; two uncomplicated pregnancies; uterine fibroid, severe menstrual pains for many years, for which she was regularly taking pain-killers, metrectomy in 2007.
History of present illness: in 2007 she developed pedal edema, outpatient work-up showed mild proteinuria and serum creatinine 150 µmol/L, which was considered as analgesic nephropathy and urinary tract infection. After metrectomy she stopped taking pain-killers, edema resolved spontaneously.
December 2014 she complained for back pain, outpatient work-up revealed serum creatinine 169 µmol/L, total calcium and urinalysis were normal. Serum electrophoresis showed total protein 93 g/L and M-spike. Spinal X-ray found osteoporosis, kidney ultrasound was unremarkable.
August 2015 her serum and urine immunochemistry revealed IgG kappa (24.6 g/L) and Bence-Jones protein kappa (207 mg/L) secretion, Bence-Jones protein in the urine was not detected. Mild hypogammaglobulinemia was found, cryoglobulines not detected, CRP and β2-microglobuline - within normal range. Urine contained traces of albumin.
January 2016 skull and pelvis X-ray and spinal MRI didn’t find any lytic changes.
March 2016 bone marrow biopsy was performed. Light microscopy showed bone marrow hypocellularity with fields of aplasia. Granulocytic lineage represented by all stages of cell maturation with predominance of mature elements and major eosinophil generations. Erythroid lineage represented by clusters of erythrokariocytes of normoblastic series. Megakaryocytes are represented by small and middle size cells with hypo- and hypersegmented normochromic nuclei. Solitary mature plasma cells dispersed interstitially, with few clusters of 3-5 cells and several small groups of lymphoid cells.
Hematologist consult ruled out the diagnosis of multiple myeloma, and patient was referred to our clinic.
At admission: conscious, alert, oriented, slightly depressed. Body temperature 36.4°C, RR 16 per minute, pulse regular 74 per minute, BP 110/60 mm Hg. Skin clear, normally colored, no edema. Eyes clear. HEENT and neck otherwise normal. Peripheral lymph node not palpable. Joints: no swelling, movements not restricted. Vertebral and flat bones percussion non painful. Lungs: clear. Heart: regular rate and rhythm, no murmur. Abdomen soft, non-tender, bowel sounds normal. Liver, spleen and kidneys not felt. Urine output 2600 ml/24 hours.
Work-up: total blood count otherwise normal, serum creatinine 213 µmol/L, total protein 90 g/l, M-spike in gamma zone. Other blood chemistry tests, serum osmolality and pH within normal range. IgA and IgM within normal range, IgG 37.4 g/L. Infectious screening negative, coagulation tests otherwise normal. Urine specific gravity 1010-1011, urine pH 5.0, proteinuria 0.5-0.01 g/24 hours, glycosuria not detected, urinary sediment unremarkable. Electrocardiogram and plain chest X-ray unremarkable. Kidneys and abdomen ultrasound – nothing but mild hepatomegaly.
Kidney biopsy
Light microscopy: sections of formalin fixed paraffin-embedded tissue were stained with H&E, Masson’s trichrome, periodic acid-Shiff and Congo red. 20 glomeruli, 4 out of them totally sclerosed (Figure 4). The rest appear enlarged, without any proliferative changes, capillary walls thin, single-contour (Figure 5). Mild interstitial fibrosis and tubular atrophy, up to 15% of parenchyma, diffuse-focal interstitial infiltration with inflammatory cells in the areas of interstitial fibrosis. Few tubules with signs of epithelial dystrophy without any casts in the tubular lumen (Figure 6). Arteries and arterioles otherwise normal. Congo red staining was negative.
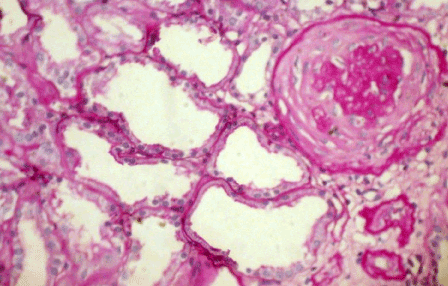
Figure 4. Case 2 - light microscopy. Totally sclerosed glomerulus. Focal tubular atrophy. PASx200
Immunofluorescence on unfixed cryo-sections: with fluorescein conjugated anti IgA, IgG, IgM, C1q, C3, fibrinogen, lambda and kappa light chains antibodies. Positive (++) staining for kappa light chains in the resorptive granules within tubular epithelial cells, and negative staining for IgG, IgA, IgM, С3, С1q, fibrinogen and lambda light chains.
Pathologists conclusion: Focal global glomerulosclerosis (20%), immunofluorescence data suggestive for light-chain tubulopathy.
Additional work-up
Bone marrow biopsy from March 2016 was re-evaluated: Iimmunohistochemistry with CD3, CD19, CD20, CD56, CD138, Cyclin D1, IgA, IgG, lambda and kappa light chains and was ordered and found large number of mature plasma cells CD138+, located inter- and para-trabecular discretely and in clusters up to 10 cells. These cells were kappa-positive and express CD56 (membrane reaction) and IgG (cytoplasmic reaction). Small B-cells (CD19+, CD20+) and small T-cells (CD3+) are dispersed. Histocytes were Cyclin D1-positive.
Clinical diagnosis: Multiple myeloma IgG kappa and Bence-Jones kappa, light-chain tubulopathy with focal global glomerulosclerosis (CKD stage 3b), secondary immunodeficiency
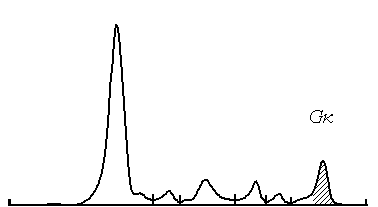
Treatment and follow-up: Patient received 4 courses of bortezomib-dexamethasone chemotherapy in our unit. At the latest evaluation October 2016 she is doing well, her serum creatinine decreased from 213 to 98 µmol/L, and immunochemistry showed the decrease of IgG-kappa secretion from 24.6 to 7.1 g/L (Figures 6 and 7), Bence-Jonce protein not detected neither in serum nor in urine. Hypogammaglobulinemia persisted.
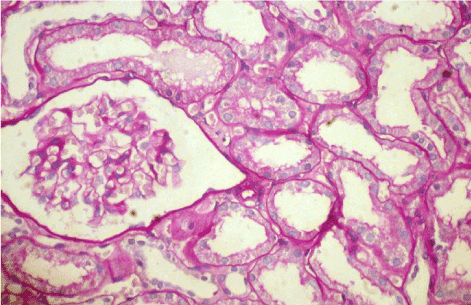
Figure 5. Case 2 - light microscopy. Otherwise normal glomerulus. Focal tubular atrophy. PASx200
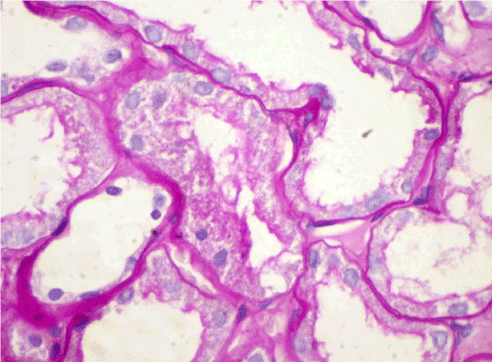
Figure 6. Case 2 - light microscopy. Tubular epithelial dystrophy. PAS x400
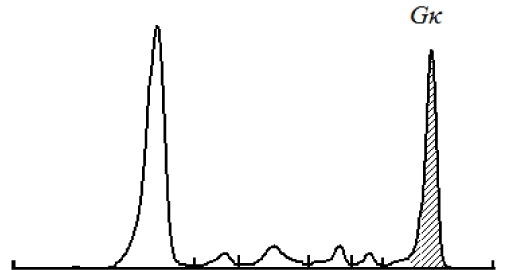
Figure 7. Case 2 - serum electrophoresis. M-spike before treatment
2021 Copyright OAT. All rights reserv
Figure 8. Case 2 - serum electrophoresis. M-spike after treatment
Final diagnosis: multiple myeloma IgG kappa and Bence-Jones kappa, light-chain tubulopathy with focal global glomerulosclerosis, AKI on the top of CKD 2, secondary immunodeficiency. Hematological and organ remission after 4 courses of chemotherapy.
Discussion
In both cases typical clinical features of MM, like ossalgia, anemia, hypercalcemia and bone lytic lesions were absent, patients presented with kidney disease, and only kidney biopsy immunopathology study gave a clue to the diagnosis. This is in agreement with the literature data, suggesting that the renal manifestations, mostly with moderate CKD and not obligatory complete FS, dominated as the clinical presentation in the majority of LCPT cases [19,24-26,29,30]. However, our cases demonstrate some peculiarities, making them rare even in the setting of LCPT, which is rare itself.
In the first case presented with unexplained new onset of severe proteinuria, routine work-up was not suggestive for myeloma: his total blood count, serum calcium, creatinine and total protein were normal, and serum electrophoresis did not show M-spike. Thus he could be suspected with non-glomerular proteinuria only because of the combination of massive proteinuria and normal serum albumin level. Therefore, kidney biopsy was performed to unmask the cause of proteinuria, and found tubular atrophy with LC lambda expression within tubular epithelial cells and very mild and unspecific focal glomerulosclerosis. That lead to the diagnosis of LCPT, and given the lack of MM signs and symptoms we considered the patient as MGRS case rather than MM. Surprisingly, his bone marrow smear showed 63.5% of plasma cells, and immunochemistry detected massive Bence-Jones protein lambda almost completely excreted in urine, which explained his normal serum protein and the absence of M-spike on standard serum electrophoresis. Severe plasma cell bone marrow infiltration, significant Bence-Jones protein secretion with biopsy-proven end-organ damage, and secondary immunodeficiency altogether fulfilled the criteria of MM. The absence of bone lytic lesions and anemia, as well as the absence of FS clinical and laboratory features in this case might be explained by the early diagnosis. The most unusual in the case 1 is the type of LC, as the association of the LCPT with lambda LC is exceptionally rare described in the literature so far.
The second case presented with 9-years history of moderate, slowly progressive renal dysfunction without significant proteinuria. Already 1.5 years before the kidney biopsy she was found with elevated total serum protein and M-spike, confirmed by detection of significant IgG kappa and mild Bence-Jones protein kappa secretion without urinary excretion of the latter. As far as she had no bone lytic lesions, anemia and hypercalcemia, and her bone marrow biopsy didn’t find plasma cell infiltration, MM diagnosis was not proved, even though she had monoclonal gammopathy, impaired kidney function and immonideficiency. She was suspected with MGRS, and kidney biopsy indeed revealed paraprotein-induced renal damage with LCPT and focal glomerulosclerosis. To differentiate MGRS and MM we ordered bone marrow iimmunohistochemistry, which provided the evidence for MM. Given FS clinical features were absent we regarded her longstanding kidney function impairment as a result of glomerulosclerosis of 20% of glomeruli secondary to the tubular damage. However, her kidney function completely recovered under hemotherapy, meaning that she had AKI on the top of her CKD, which is most unusual. Our interpretation is that the patient had a long course of smoldering myeloma with LCPT and recent evolution towards MM with exacerbation of kidney damage. The latter is suggestive for cast-nephropathy rather than for LCPT, but tubular casts were not found in her biopsy. There were no other reasons for AKI beyond LCPT, as she was not taking analgesics for many years before biopsy, didn’t receive contrast-media, antibiotics or any other nephrotoxic agents and had no signs of volume depletion.
Finally, in both cases treatment with the bortezomib-based chemotherapy regimens was successful and lead to hematologic and organ remission.
Conclusions
Light-chain proximal tubulopathy is an infrequent variant of paraproteinemic kidney damage in patients mostly not demonstrating clinical features of multiple myeloma. This lesion is almost always induced by kappa light chains, presenting with moderate CKD, and, rarely, with Fanconi syndrome. None of our two patients met multiple myeloma criteria at admission, and only kidney biopsy, which discovered light-chain proximal tubulopathy, prompted further work-up in turn resulted in the diagnosis of multiple myeloma. Case one is an extremely rare example of light chain lambda deposition in the proximal tubules, case two demonstrates acute kidney injury on the top of chronic kidney disease, unusual for the light-chain proximal tubulopathy. We conclude that kidney biopsy should be performed, unless contraindicated, in all patients with proteinuria and/or impaired kidney function of unknown origin; and that the chemotherapy with bortezomib-based regimens is effective for treatment of myeloma with light-chain proximal tubulopathy.
Acknowledgments
We have to thank doctors Alla Kovrigina, Eugeny Filimonov and Nuriett Khuajeva for their help in the diagnostics and treatment of our patients.
References
- Rajkumar SV, Kyle RA (2005) Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 80: 1371-1382. [Crossref]
- International Myeloma Working Group (2003) Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br J Haematol 121: 749-757. [Crossref]
- Haas M, Spargo BH, Wit EJ, Meehan SM (2000) Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis 35: 433-447. [Crossref]
- Korbet SM, Schwartz MM (2006) Multiple myeloma. J Am Soc Nephrol 17: 2533-2545. [Crossref]
- DeFronzo RA, Cooke CR, Wright JR, Humphrey RL (1978) Renal function in patients with multiple myeloma. Medicine (Baltimore) 57: 151-166. [Crossref]
- Ganeval D, Rabian C, Guérin V, Pertuiset N, Landais P, et al. (1992) Treatment of multiple myeloma with renal involvement. Adv Nephrol Necker Hosp 21: 347-370. [Crossref]
- Winearls CG (1995) Acute myeloma kidney. Kidney Int 48: 1347-1361. [Crossref]
- Clark AD, Shetty A, Soutar R (1999) Renal failure and multiple myeloma: pathogenesis and treatment of renal failure and management of underlying myeloma. Blood Rev 13: 79-90. [Crossref]
- Sanders PW, Booker BB (1992) Pathobiology of cast nephropathy from human Bence Jones proteins. J Clin Invest 89: 630-639. [Crossref]
- Cohen AH (1998) The kidney in plasma cell dyscrasias: Bence-Jones cast nephropathy and light chain deposit disease. Am J Kidney Dis 32: 529-532. [Crossref]
- Ronco PM (1999) Paraneoplastic glomerulopathies: new insights into an old entity. Kidney Int 56: 355-377. [Crossref]
- Markowitz GS (2004) Dysproteinemia and the kidney. Adv Anat Pathol 11: 49-63. [Crossref]
- Herrera GA, Joseph L, Gu X, Hough A, Barlogie B (2004) Renal pathologic spectrum in an autopsy series of patients with plasma cell dyscrasia. Arch Pathol Lab Med 128: 875-879. [Crossref]
- Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, et al (2004). Proliferative glomerulonephritis with monoclonal IgG deposits; a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int 65: 85-96. [Crossref]
- Merlini G, Pozzi C (2007) Mechanisms of renal damage in plasma cell dyscrasias: an overview. Contrib Nephrol 153: 66-86. [Crossref]
- Ronco P, Plaisier E, Aucouturier P (2010) Ig-related renal disease in lymphoplasmacytic disorders: an update. Semin Nephrol 30: 557-569. [Crossref]
- Heher EC, Goes NB, Spitzer TR, Raje NS, Humphreys BD, et al. (2010) Kidney disease associated with plasma cell dyscrasias. Blood 116: 1397-1404. [Crossref]
- Cambier JF, Ronco P (2012) Onco-nephrology: glomerular diseases with cancer. Clin J Am Soc Nephrol 7: 1701-1712. [Crossref]
- Maldonado JE, Velosa JA, Kyle RA, Wagoner RD, Holley KE, et al. (1975) Fanconi syndrome in adults. A manifestation of a latent form of myeloma. Am J Med 58: 354-364. [Crossref]
- Tubbs RR, Gephardt GN, McMahon JT, Hall PM, Valenzuela R, et al. (1981) Light chain nephropathy. Am J Med 71: 263-269. [Crossref]
- Zakharova EV, Fedorova ND, Ratner MI (1983) Fanconi syndrome in adults (case of prolonged course of the Fanconi syndrome in non-malignant monoclonal gammopathy). Terapevticheskii arkhiv 10: 109-113.
- Confalonieri R, Barbiano di Belgiojoso G, Banfi G, Ferrario F, Bertani T, et al. (1988) Light chain nephropathy: histological and clinical aspects in 15 cases. Nephrol Dial Transplant 3: 150-156. [Crossref]
- Bate KL, Clouston D, Packham D, Ratnaike S, Ebeling PR (1998) Lambda light chain induced nephropathy: a rare cause of the Fanconi syndrome and severe osteomalacia. Am J Kidney Dis 32: E3. [Crossref]
- Lacy MQ, Gertz MA (1999) Acquired Fanconi's syndrome associated with monoclonal gammopathies. Hematol Oncol Clin North Am 13: 1273-1280. [Crossref]
- Messiaen T, Deret S, Mougenot B, Bridoux F, Dequiedt P, et al (2000). Adult Fanconi syndrome secondary to light chain gammopathy. Clinicopathologic heterogeneity and unusual features in 11 patients. Medicine (Baltimore) 79: 135-154. [Crossref]
- Ma CX, Lacy MQ, Rompala JF, Dispenzieri A, Rajkumar SV, et al (2004) Acquired Fanconi syndrome is an indolent disorder in the absence of overt multiple myeloma. Blood 104: 40-42. [Crossref]
- Batuman V (2007) Proximal tubular injury in myeloma. Contrib Nephrol 153: 87-104. [Crossref]
- Larsen CP, Bell JM, Harris AA, Messias NC, Wang YH, et al. (2011) The morphologic spectrum and clinical significance of light chain proximal tubulopathy with and without crystal formation. Mod Pathol 24: 1462-1469. [Crossref]
- Sharma SG, Bonsib SM, Portilla D, Shukla A,Woodruff AB, et al. (2012) Light Chain Proximal Tubulopathy: Expanding the Pathologic Spectrum with and without Deposition of Crystalline Inclusions. ISRN Pathology: 541075.
- Leung N, Bridoux F, Hutchison CA, Nasr SH, Cockwell P, et al. (2012) International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 120: 4292-4295. [Crossref]
- Fermand JP, Bridoux F, Kyle RA, Kastritis E, Weiss BM, et al. (2013) How I treat monoclonal gammopathy of renal significance (MGRS). Blood 122: 3583-3590. [Crossref]
- Ludwig H, Adam Z, Hajek R, Greil R, To´thova E, et al (2010) Light Chain–Induced Acute Renal Failure Can Be Reversed by Bortezomib-Doxorubicin-Dexamethasone in Multiple Myeloma: Results of a Phase II Study. J Clin Oncol 28: 4625-4641. [Crossref]