Abstract
Background: Endometrial cancer rates have risen sharply in the past decade and are projected to continue increasing. With few treatment options available, this study examines a novel proteosome inhibitor UR238 as a potential new therapy for endometrial cancer.
Methods: UR238 is a 3rd generation proteosome inhibitor. Expression of proteasome subunits PSMB5 and LMP7, putative targets of UR238, were assessed through the Human Protein Atlas in uterine cancer patients. Functional assays, RT-PCR and western blotting were used to determine UR238’s efficacy, induction of endoplasmic reticular (ER) stress, autophagy, and apoptosis. An AN3CA xenograft mouse model was used to assess the in vivo effects of UR238.
Results: LMP7 is increased in endometrial cancer tumors. UR238 exhibits inhibition of chymotrypsin-like proteasomal activity and compared to known proteosome inhibitors, UR238 enhances PARP cleavage. Endometrial cancer cell lines treated with UR238 demonstrate reduced proliferation and increased markers of ER stress and other key markers of the apoptotic pathway. AN3CA expressing xenograft mice treated with UR238 demonstrate significant reduction in mean tumor volumes compared with vehicle control. Lastly, UR238 downregulates HE4 expression which appears to be independent of unfolded protein response (UPR) signaling.
Conclusion: UR238 inhibits cell proliferation in endometrial cancer cell lines and exhibits antitumorigenic effects in xenograft mouse models. UR238 induces ER stress, triggers UPR signaling, and triggers autophagy. UR238 also downregulates HE4 expression although, this is likely independent of UPR signaling. Proteosome inhibitors demonstrate promising activity in endometrial cancer and should be further investigated in these malignancies.
Keywords
UR238, endometrial cancer, proteasome inhibitor, endoplasmic reticulum stress
Introduction
In the United States, 69,000 women are diagnosed with endometrial cancer (EC) annually and by 2050, the incidence is expected to increase to 120,000 cases annually [1,2]. Among women with newly diagnosed EC, ~30% will be advanced stage disease necessitating treatment with systemic therapy [3]. In recent years, newer therapies such as checkpoint inhibitors [4,5] and targeting the HER2 receptor have improved treatment responses [6,7], however, there is still an unmet need for systemic treatment options for advanced and recurrent EC patients whose overall 5-year survival is less than 20% [8]. The use of proteasome inhibitors has not been widely studied in EC.
In this study, our aim was to establish proteasomal function as a therapeutic target in EC given the known upregulation of proteasome activity in EC [9]. Proteosome inhibitors bortezomib and carfilzomib have been utilized in the treatment of non-solid malignancies, largely multiple myeloma where they have shown positive impacts on patient outcomes and survival [10,11]. Previous studies of bortezomib in solid malignancies have not shown the same promising outcomes; Huang, et al. [12], reviewed clinical trials investigating bortezomib in the treatment of solid malignancies and there was a lack of therapeutic effect observed when the drug was used as a single agent or in combination with other agents [12]. It has been theorized that bortezomib may have poor penetration into solid tumor compared to its availability in blood/bone marrow contributing to its poor performance in solid malignancies [13]. The ClogP value of a drug is reflective of its lipophilicity and water solubility which translates into its absorption and solubility [14]. Drugs with a suboptimal ClogP values have poor solubility and thereby limited therapeutic utility in solid tumors. UR238 was medicinally optimized and developed from current proteosome inhibitor chemical structures with the intent to improve ClogP and other medicinal chemistry properties. The improved CLogP of UR238 increases the drugs penetration into solid tumors compared with the earlier generations of proteasome inhibitors.
Here we investigate the 3rd generation proteasome inhibitor, UR238, and its effect on endoplasmic reticular (ER) stress. ER stress initially induces the unfolded protein response (UPR) pathway, which can serve to maintain cellular homeostasis in an afflicted cell. However, during periods of prolonged or severe ER stress, the UPR pathway shifts to promoting cell death. Given that ER stress and UPR activation have been demonstrated in EC and linked to chemoresistance [15,16], inducing ER stress further beyond a cells adaptive limits may have therapeutic potential in treatment of EC.
We will also investigate how UR238 and UPR signaling affect the expression of Human Epididymis Protein 4 (HE4) produced through the expression of the WFDC2 gene. HE4 is known to enhance EC proliferation, growth, and invasion [17,18]. Additionally, HE4 overexpression has been shown to upregulate immune checkpoint PD-L1, and promote a suppressive immune microenvironment, together indicating HE4 as a promising therapeutic target in EC [19]. We have shown that HE4 expression is partially maintained though proteasome regulation. Here, we demonstrate that both UR238 treatment and ER stress suppress HE4 expression, and we further examine how each condition influences HE4 through UPR signaling. Additionally, this study demonstrates that UR238 is more potent than the current proteosome inhibitors in clinical use.
Materials and methods
Cell culture and materials: The human endometrial cancer cell lines ECC-1 (RPMI), HEC1A (McCoy’s 5A), MFE296 (MEM), KLE (DMEM:F-12), AN3CA (MEM), and MFE280 (MEM) all of which express HE4 were obtained as previously described [20]. Each cell line was cultured at 37 °C with 5% CO2 in its respective medium (specified in brackets), supplemented with 10% FBS. UR238 was synthesized and the structure and purity (>95%) were confirmed using nuclear magnetic resonance and high-performance liquid chromatography. A 1000x stock solution was prepared in dimethyl sulfoxide, stored at -20 °C, and mixed with cell culture medium prior to cell treatment. All other materials were sourced from vendors as follows; Chemicals: Bafilomycin (Cell Signaling Technology, Cat. No. 54645), Palmitic acid (as in a complex with BSA, Cayman, Cat. No. 29558), BSA-control (Cayman, Cat. No. 29556), Thapsigargin (Cayman, Cat. No. 10522), Tunicamycin (Cell Signaling Technology, Cat. No. 12819), 4μ8C (Cayman, Cat. No. 22110), GSK2606414 (MCE, Cat. No. HY-18072); siRNAs: Silencer Select siRNAs (ThermoFisher) for control (Cat. No. 4390843), CHOP (ID No. s3997), ATF6 (ID No. s22688) and ATF4 (ID No. s1702). Antibodies: Nrf2 (Abcam, Cat. No. ab62352), LC3 (MBL, Cat. No. PM036), ATF6 (BioLegend, Cat. No. 853101), ATF4 (BioLegend, Cat. No. 693901). All other antibodies were purchased from Cell Signaling Technology: Ubiquitin (Cat. No. 3933), β-Actin (Cat. No. 3700), BiP (Cat. No. 3177), CHOP (Cat. No. 2895), XBP-1s (Cat. No. 12782), P-p38 (Cat. No. 4511), p38 (Cat. No. 9212), P-JNK (Cat. No. 4668), JNK (Cat. No. 9252), P-Erk1/2 (Cat. No. 4370), Erk1/2 (Cat. No. 9102), p21 (Cat. No. 2947), p27 (Cat. No. 3686), α-Tubulin (Cat. No. 2144), Cleaved PARP (Cat. No. 5625), GADD34 (Cat. No. 41222).
Proteasome assay: Chymotrypsin-like, caspase-like, and trypsin-like proteases activities were measured using the Proteasome-Glo Cell-Based Assay (Promega, Cat. No. G8660, G8760, G8860) according to the manufacturer's recommendations. Briefly, ECC-1 cells were harvested by trypsinization and were washed thoroughly with complete medium to remove contaminating trypsin or chymotrypsin carryover. The cells were then plated into a 96-well white plate and allowed to adhere overnight. Following this, the cells were treated with various concentrations of UR238 for 2 hours. They were then permeabilized using the buffer provided in the kit, which contains specific luminogenic peptide substrates: Suc-LLVY-, Z-LRR- or Z-nLPnLD-aminoluciferin for the chymotrypsin-like, trypsin-like or caspase-like activities, respectively. Upon cleavage by the proteasome, aminoluciferin is released, enabling the luciferase reaction. Luminescence signal was measured using a Synergy 2 (BioTek) microplate reader.
Cell transfection, immunoblot analysis, and measurement of HE4 Levels: Transfection and immunoblot analyses were conducted as previously described [21]. HE4 levels were measured using the Human HE4/WFDC2 DuoSet enzyme-linked immunosorbent assay (R&D Systems, Cat. no. DY6274-05), following the manufacturer's protocol. For each reaction, 2 μg of ECC-1 cell extracts were used.
Measurement of cell population and apoptosis: Cell population was measured using the Sulforhodamine B (SRB) assay [22]. Apoptotic cells were measured by evaluating caspase-3/7 activities in cell culture, utilizing the Caspase-Glo 3/7 Assay (Promega, Cat. No. G8091). This assay includes a luminogenic peptide substrate, DEVD, specific for caspase-3/7 activity.
Polymerase Chain Reaction (PCR): ECC-1 cells were treated with UR238 (250 nM) for 24 hours. RNA isolation and cDNA synthesis were conducted as previously described [21]. Quantitative real-time PCR was conducted using a QuantStudio 12K Flex System (ThermoFisher) with Power SYBR Green PCR Master Mix (ThermoFisher, Cat. No. 4367659) with primers as follows: GAPDH (forward: AGC CAC ATC GCT CAG ACA C, reverse: GCC CAA TAC GAC CAA ATC C); usXBP1 (forward: CAG CAC TCA GAC TAC GTG CA, reverse: ATC CAT GGG GAG ATG TTC TGG; sXBP1 (forward: CTG AGT CCG AAT CAG GTG CAG, reverse: ATC CAT GGG GAG ATG TTC TGG); CHOP (forward: AGA ACC AGG AAA CGG AAA CAG A, reverse: TCT CCT TCA TGC GCT GCT TT); BiP (Forward: TGT TCA ACC AAT TAT CAG CAA ACT C, reverse: TTC TGC TGT ATC CTC TTC ACC AGT).
Results
Endometrial cancer tissue exhibits increased levels of LMP7, a primary binding target of proteosome inhibitors
The PSMB5 and LMP7 genes encode 20S and 20Si proteasome subunits (i.e. β5 and β5i) and are the primary targets of proteosome inhibitors [23]. Microarray analysis of PMBS5 and LMP7 was performed using the Human Protein Atlas. Normalized relative protein expression (NRPX) is calculated as log2 (intensity), derived from global protein abundance measurements at the protein level. These measurements were obtained through mass spectrometry experiments performed by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) on tissues using isobaric tandem mass tags (TMT). Compared to non-malignant tissues, endometroid endometrial cancer tissue exhibits increased expression of LMP7 (Figure 1).
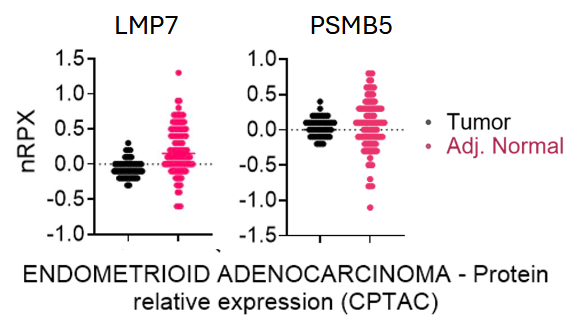
Figure 1. Figure generated from The Human Protein Atlas; normalized relative protein expression (NRPX) of LMP7 and PSMB5 in benign tissue specimens vs. tumor specimens from patients with endometrial cancer (Unpaired p-value: <0.0001 and 0.6440, respectively). NRPX is calculated as log2(intensity), derived from global protein abundance measurements at the protein level. These measurements were obtained through mass spectrometry experiments performed by CPTAC on tissues using isobaric tandem mass tags (TMT)
UR238 more effectively reduces endometrial cancer cell proliferation and enhances PARP Cleavage compared to Carfilzomib
We compared UR 238’s ability to reduce endometrial cancer cell population to that of Carfilzomib. After 72 hours of treatment, UR238 more effectively reduced ECC-1 cell numbers at both 125 and 250 nM compared to Carfilzomib (Figure 2A). Treatment of various cancer cell lines with Carfilzomib has previously been shown to result in cleavage of poly (ADP-ribose) polymerase (PARP), a key protein involved in the DNA damage repair pathway, and its cleavage is a well-known indicator of the activation of cellular apoptosis [24]. We compared PARP cleavage in ECC-1 cells treated with varying concentrations of UR238 vs. Carfilzomib (0, 250, 500 and 750 nM). Immunoblot analysis demonstrated UR238 enhanced PARP cleavage compared to Carfilzomib at all concentrations (Figure 2B); this finding was further supported by increased ubiquitinated protein accumulation in UR238 treated cells compared Carfilzomib (Figure 2C).
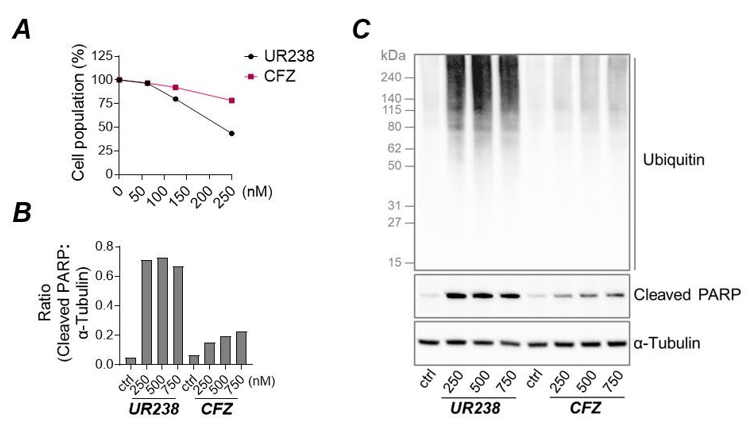
Figure 2: (A) ECC-1 cells were treated with a range of concentrations of UR238 and Carfilzomib for 72 hours. After treatment, cells were fixed, and cell population was measured by the sulforhodamine B (SRB) assay. (B,C) ECC-1 cells were treated for 48 hours with varying concentrations of either UR238 or Carfilzomib. Following treatment, protein extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting to detect PARP cleavage and protein ubiquitination. α-tubulin served as a loading control. Densitometric analysis of cleaved PARP normalized to α-tubulin is shown in (B)
UR238 inhibits proteasome activity and reduces endometrial cancer cell proliferation
We evaluated the effects of UR238 on endometrial cancer cell lines by treating each line with various concentrations of UR238 for 24 and 48 hours. Within the first 24 hours, UR238 treatment only moderately affected cell populations, even at the highest concentration tested (1000 nM), with 19%-41% inhibition compared to the control. ECC-1, HEC1A, and AN3CA cell lines were more sensitive, showing 37%, 41%, and 40% inhibition, respectively, while KLE showed only a 19% reduction. After 48 hours, the effects were more pronounced with UR238. ECC-1 cells treated with UR238 (1000 nM) showed an 82% reduction. Next, based on its chemical structure we predict that the proteasome is a target of UR238. To evaluate the selectivity and efficacy of UR238 toward three major proteolytic activities within the proteasome—chymotrypsin-like, trypsin-like, and caspase-like (or post-glutamyl peptide hydrolytic) activities—ECC-1 cells were treated with various concentrations of UR238 (0.001-10 μM) and monitored for the cleavage of a luminogenic peptide substrate over 2 hours (Figure 3A and 3B). UR238 treatment in ECC-1 cells exhibited significant inhibition of chymotrypsin-like activity (IC50 = 0.039 μM; Figure 3C) in a dose-dependent manner, with much lesser inhibitory activities against caspase-like and trypsin-like activities (IC50 = 0.71 and 1.15 μM, respectively). When the proteasome is inhibited, it loses its ability to degrade ubiquitinated proteins, resulting in their accumulation within cells. To confirm that UR238 works through the proteasome pathway, we treated cells with UR238, which led to a significant accumulation of ubiquitinated proteins (Figure 3D).
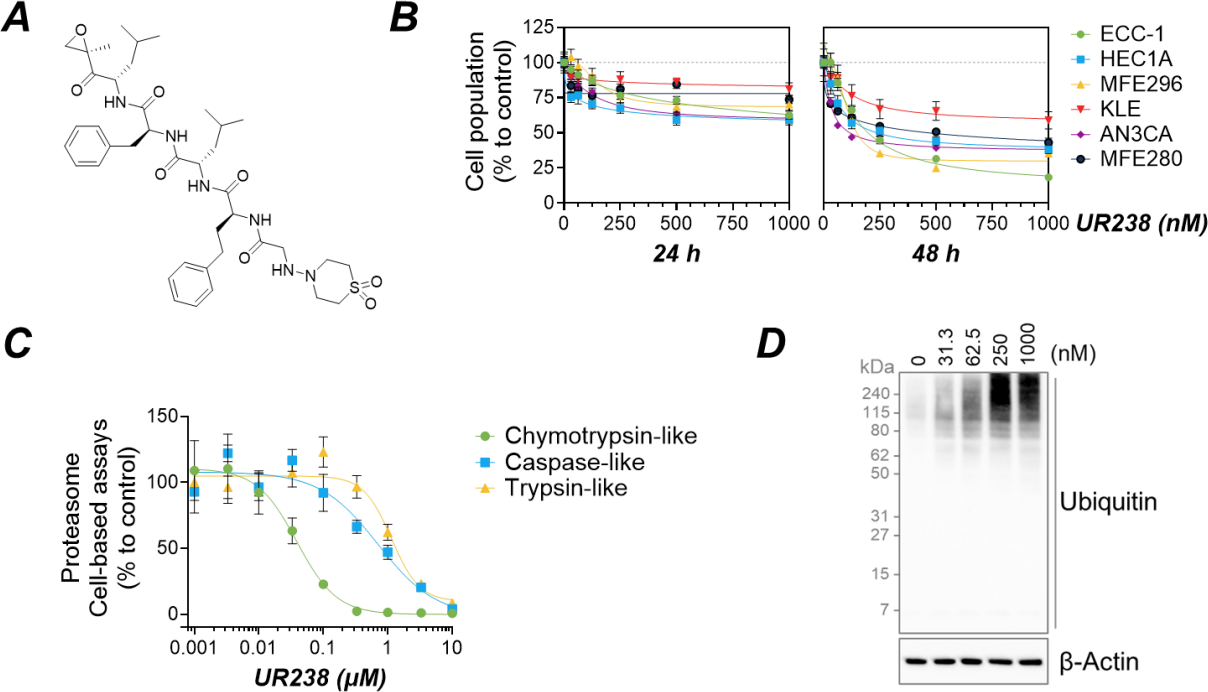
Figure 3. (A) Chemical structure of UR238. (B) Six different endometrial cancer cell lines were treated with a range of concentrations of UR238 for 24 or 48 hours. After treatment, cells were fixed, and cell population was measured by SRB assay. (Mean ± SEM, N=6). (C) ECC-1 endometrial cancer cells were treated with various concentrations of UR238 for 2 hours. Following treatment, the cells were permeabilized. Chymotrypsin-like, caspase-like, and trypsin-like proteases activities were then measured using specific luminogenic substrates, as detailed in “Materials and Methods”. (D) ECC-1 cells were treated with the indicated concentrations of UR238 for 24 hours. After treatment, protein extracts were prepared, separated by SDS-PAGE electrophoresis, and then immunoblotted to detect ubiquitinylated proteins, with β-actin used as a loading control
UR238 induces ER stress and activates UPR pathways in endometrial cancer cells
The inhibition of proteasome activity is known to enhance endoplasmic reticulum (ER) stress in cells, triggering the activation of a conserved signaling cascade known as the unfolded protein response (UPR) pathway. In Figure 4A, we analyzed the expression of UPR pathway upon UR238 treatment along with other proteins known to be affected by the ER stress or proteasome inhibition. First, we observed that UR238 treatment in ECC-1 cells significantly upregulated the expression of BiP, a key regulator of the UPR. The UPR signaling is mediated by three main pathways: IRE1α, PERK, and ATF6. UR238 was found to activate all three pathways, as evidenced by the upregulation of XBP-1s (the spliced isoform of XBP1), CHOP, and cleaved ATF6, which are representative markers of the IRE1α, PERK, and ATF6 pathways, respectively. In agreement with this, quantitative reverse transcription PCR (qRT-PCR) analysis revealed that UR238 transcriptionally regulates UPR signaling (Figure 4B). UR238 also activated MAPK pathways (p38, Erk1/2, JNK) and redox sensor Nrf2, both linked to ER stress and UPR [24]. Autophagy is a vital cell survival mechanism that helps cells manage stressors like oxidative stress, nutrient deprivation, and ER stress [25]. LC3 is a key marker for monitoring this process. Our study showed that UR238 clearly induces LC3. Moreover, UR238 treatment upregulated the cell-cycle inhibitors p21 and p27, as seen by other proteasome inhibition [26,27]. These effects were not limited to ECC-1 cells, as similar results were observed in five additional endometrial cancer cell lines (Figure 4A and 4C).

Figure 4. (A) ECC-1 cells were treated with UR238 at the concentrations and durations as indicated. Following treatment, protein extracts were prepared, separated by SDS-PAGE electrophoresis, and immunoblotted with the indicated specific antibodies. α-tubulin was used as a loading control. (B) ECC-1 cells were treated with UR238 (250 nM) for 24 hours. The cells were lysed, and isolated RNAs were analyzed for the indicated genes via quantitative reverse transcription PCR (qRT-PCR), as described in “Materials and Methods”. sXBP1: spliced XBP1; usXBP1: unspliced XBP1. (C) The same as in (A), but the listed cell lines were incubated with UR238 for 24 hours at 1000 nM
UR238 Induces apoptosis and potential synergy through autophagy and CHOP signaling
Severe or prolonged ER stress can lead to cell death, typically via apoptosis [28]. We incubated KLE and ECC-1 cells with various concentrations of UR238 for 48 hours and measured caspase-3/7 activities, key mediators of the final apoptotic steps. We found that UR238 induces apoptosis in both KLE and ECC-1 cells in a dose-dependent manner (Figure 5A). ECC-1 cells exhibited greater sensitivity in cleaving a pro-luminescent caspase-3/7 DEVD-substrate, with a 41-fold increase in apoptotic cells at 1000 nM compared to the control, whereas KLE cells showed only a 9-fold increase. Next, we assess for autophagy which can both protect cells or promote cell death under ER stress [29]. To investigate the role of autophagy upon UR238 treatment, we used bafilomycin A1, a potent V-ATPase inhibitor to block lysosomal degradation and thereby disrupt autophagic flux [30]. Bafilomycin A1 treatment led to a greater increase in LC3 expression than UR238 (Figure 5B) and significantly enhanced apoptosis (Figure 5B and 5C), suggesting that autophagy induction by UR238 promotes cell survival, and its inhibition can potentiate the therapeutic effects of UR238. Histone deacetylase (HDAC) inhibitors are known to induce autophagy [31]. The clinical agent vorinostat, or SAHA, has demonstrated a synergistic anticancer effect when combined with proteasome inhibitors [32]. Therefore, we tested whether UR238 has similar activity and found that vorinostat enhanced UR238’s effects in ECC-1 cells (Figure 5D). The Bcl-2 family proteins regulate cell survival and death through anti-apoptotic (e.g., Bcl-2, Mcl-1) and pro-apoptotic (e.g., Bax, Bim) members. While inducing apoptosis, we observed that UR238 upregulated Mcl-1 expressions in several endometrial cancer cell lines (Figure 5E and 5F), suggesting a compensatory survival mechanism. Since doxorubicin, a standard treatment for endometrial cancer, is known to suppress Mcl-1, we hypothesized that combining it with UR238 could enhance therapeutic efficacy [33]. Indeed, doxorubicin reduced UR238-induced Mcl-1 expression and simultaneously increased apoptosis (Figure 5G and 5H). Similarly, co-treatment with A1210477, a selective Mcl-1 inhibitor, yielded comparable results (Figure 5I), supporting the potential of UR238 in combination therapy. Lastly, CHOP induces apoptosis during severe ER stress in UPR signaling [34,35]. To confirm that CHOP induction by UR238 follows its previously established role, we knocked down CHOP expression in ECC-1 cells using siRNAs and treated them with UR238. Our data showed that CHOP knockdown induced resistance to UR238 (1000 nM) by 31%, suggesting that UR238 exerts CHOP-mediated cytotoxic effects (Figure 5J).
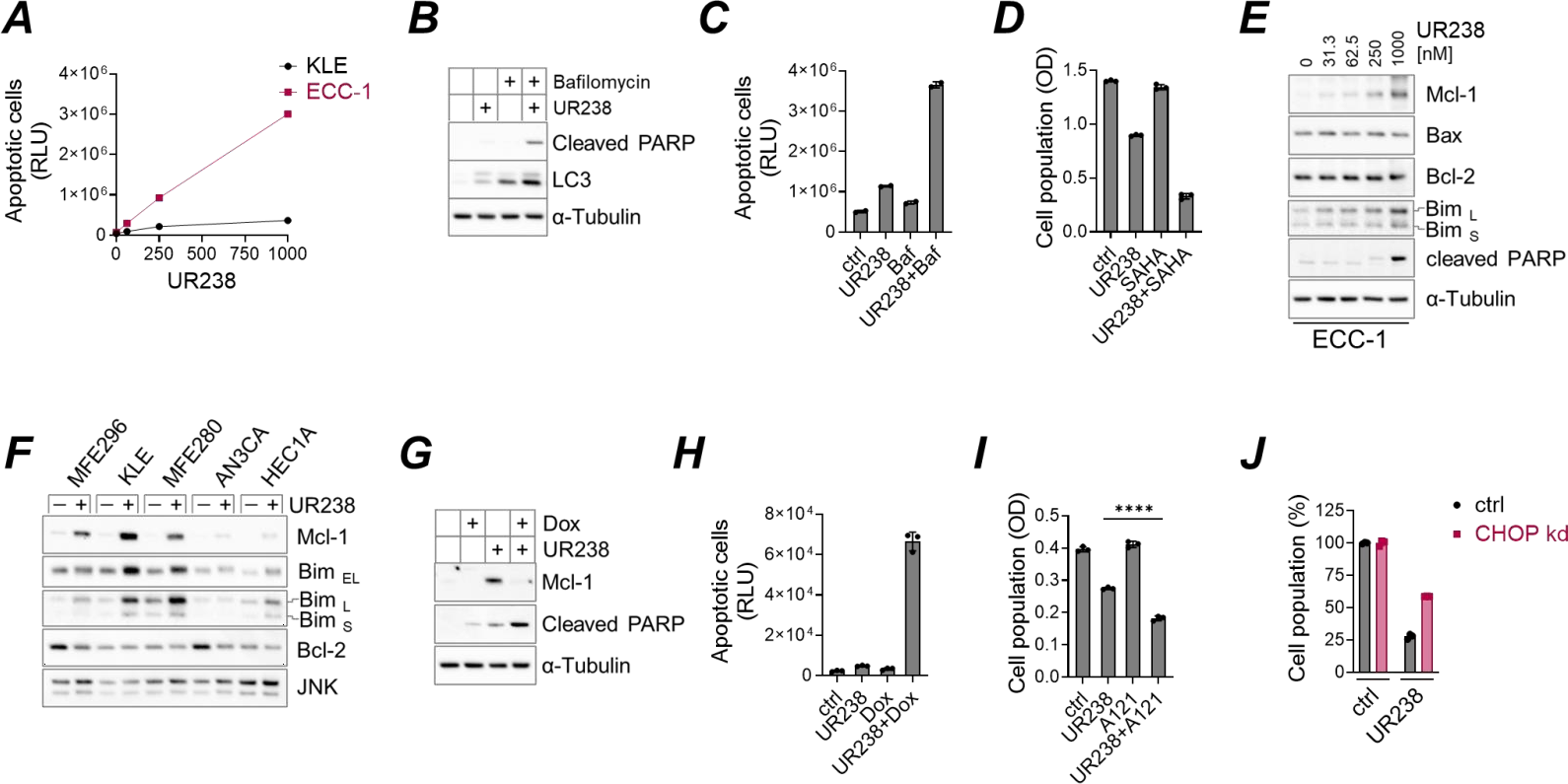
Figure 5. (A) KLE and ECC-1 cells were treated with UR238 (nM) for 48 hours. Apoptotic cells were then measured using luminogenic substrates sensitive to caspase-3/7 activity, as detailed in 'Materials and Methods'. The results were normalized to the cell population for each condition as determined by the SRB assay. (B) ECC-1 cells were incubated with UR238 (250 nM) with or without bafilomycin A1 (100 nM) for 24 hours. After treatment, immunoblotting was performed to detect markers for apoptosis (cleaved PARP) and autophagy (LC3), using α-tubulin as a loading control. (C) ECC-1 cells were treated with UR238 (250 nM), bafilomycin A1 (300 nM), or a combination of both for 24 hours. Apoptotic cells were then measured as described in (A). (D) ECC-1 cells were treated with UR238 (500 nM for 48 hours), vorinostat (SAHA, 20 µM for 24 hours), or a combination of both. Cell population was assessed using the SRB assay. (E, F) Endometrial cancer cell lines were treated with UR238 at the indicated concentrations (E) or at 1000 nM (F) for 24 hours. Following treatment, immunoblotting was performed using the specified antibodies. (G,H) ECC-1 cells were treated with UR238 (250 nM for H, 1000 nM for G), doxorubicin (5 µM), or a combination of both for 24 hours. Protein extracts were then prepared and subjected to immunoblotting using the indicated antibodies (G), or apoptotic cells were quantified as shown in (A). (I) ECC-1 cells were treated with UR238 (250 nM) or the Mcl-1 inhibitor A1210477 (10 µM) for 24 hours. Cell population was assessed using the SRB assay. ****: <0.0001. (J) ECC-1 cells were transiently transfected with siRNA against CHOP or with a non-targeting control for 48 hours. UR238 was added for the last 24 hours. Cell populations were measured by the SRB assay
Effect of UR238 on AN3CA endometrial cancer cell xenografts growing in NSG mice
Using proteomic data retrieved from cProSite [36], we observed a significant upregulation of ER stress markers in endometrial tumors. BiP, PERK, and ATF6 were markedly upregulated in tumor tissues. Compared to normal tissues (Figure 6A; p < 0.0001), whereas IRE1α expression showed no significant difference (p = 0.7177). These findings suggest that targeting ER stress pathways may represent a promising therapeutic strategy for endometrial cancer. Next, we conducted the in vivo evaluation of UR238. AN3CA, a cell line derived from human endometrial adenocarcinoma, was cultured and injected subcutaneously (1.0 x 105 cells) into NSG mice to create AN3CA tumor xenografts. A total of 10 mice were used in the study. On day 9 when all mice had measurable subcutaneous tumors, mice were treated with either vehicle (water+ 40% HBC+ Kolliphor=14:5:1) or UR238 (5mg/kg) three times per week. Tumor volume was measured on days 9, 13, 17 and 20; mice were sacked on day 20. A repeated measures analysis of variance was performed using maximum likelihood estimation with group, day, and the interaction between group and day as fixed effects. The correlation of repeated measures on the same subject over time was handled using an unstructured covariance which was allowed to vary by treatment condition. Model assumptions were verified graphically. Analysis was conducted using SAS v9.4 Proc Mixed. We found significant differences between the groups at days 13, 17, 19 with respect to tumor volume, where UR238 treated group has a smaller average tumor volume compared to vehicle (p values by respective day: Day 13= 0.0108, Day 17 = 0.0378, Day 20= 0.0109.) (Figure 6B).

Figure 6. (A) BiP, ATF6 and PERK protein abundance between tumors and normal adjacent tissues. (Unpaired p-value: <0.0001) (B) AN3CA cells derived from human endometrial adenocarcinoma were injected into NSG mice to create tumor xenografts. Mice were treated with either a vehicle or UR238 (5 mg/kg) three times per week. Tumor volumes were measured on days 9, 13, 17, and 20. Statistical analysis revealed that UR238 significantly reduced tumor volume compared to the vehicle at days 13, 17, and 20 (p<0.05)
UR238 and ER Stress reduce HE4 levels in endometrial cancer cells
HE4, a protein overexpressed in endometrial cancer [37], is a potential therapeutic target for cancer treatment [38]. In our efforts to identify an effective HE4 inhibitor, we previously conducted a drug library screening of pre-existing bioactive compounds with established molecular targets. This screening identified proteasome inhibition as a viable option to inhibit HE4. Notably, treatment with UR238 significantly decreased HE4 levels in ECC-1 cells. Additionally, severe ER stress induced by thapsigargin and tunicamycin treatment (Th+Tu hereafter; Figure 7A) also reduced HE4 levels (Figure 7B). When UPR is activated, it affects protein synthesis and gene expression [39]. Thus, we investigated whether UPR signaling is responsible for HE4 inhibition upon ER stress. We used specific pharmacologic inhibitors against IRE1α and PERK (4μ8C and GSK2606414, respectively) and ATF6 knockdown ECC-1 cells. Treatment with 4μ8C and GSK2606414 effectively suppressed the downstream targets of IRE1α and PERK (XBP-1s and ATF4/CHOP, respectively). ATF6 was clearly downregulated in ATF6 knockdown cells (Figure 7C and 7D). We found that inhibiting PERK, but not IRE1α or ATF6, restored HE4 levels that had been reduced by Th+Tu treatment, to levels comparable to untreated controls (Figure 7B). qRT-PCR analysis also showed that PERK inhibition partially restored HE4 levels transcriptionally (Figure 7F). Interestingly, inhibiting PERK signaling by GSK2606414 or through ATF4/CHOP knockdown failed to restore HE4 reduced by UR238 (Figure 7B and 7E), suggesting that UR238 regulates HE4 independently of PERK signaling.
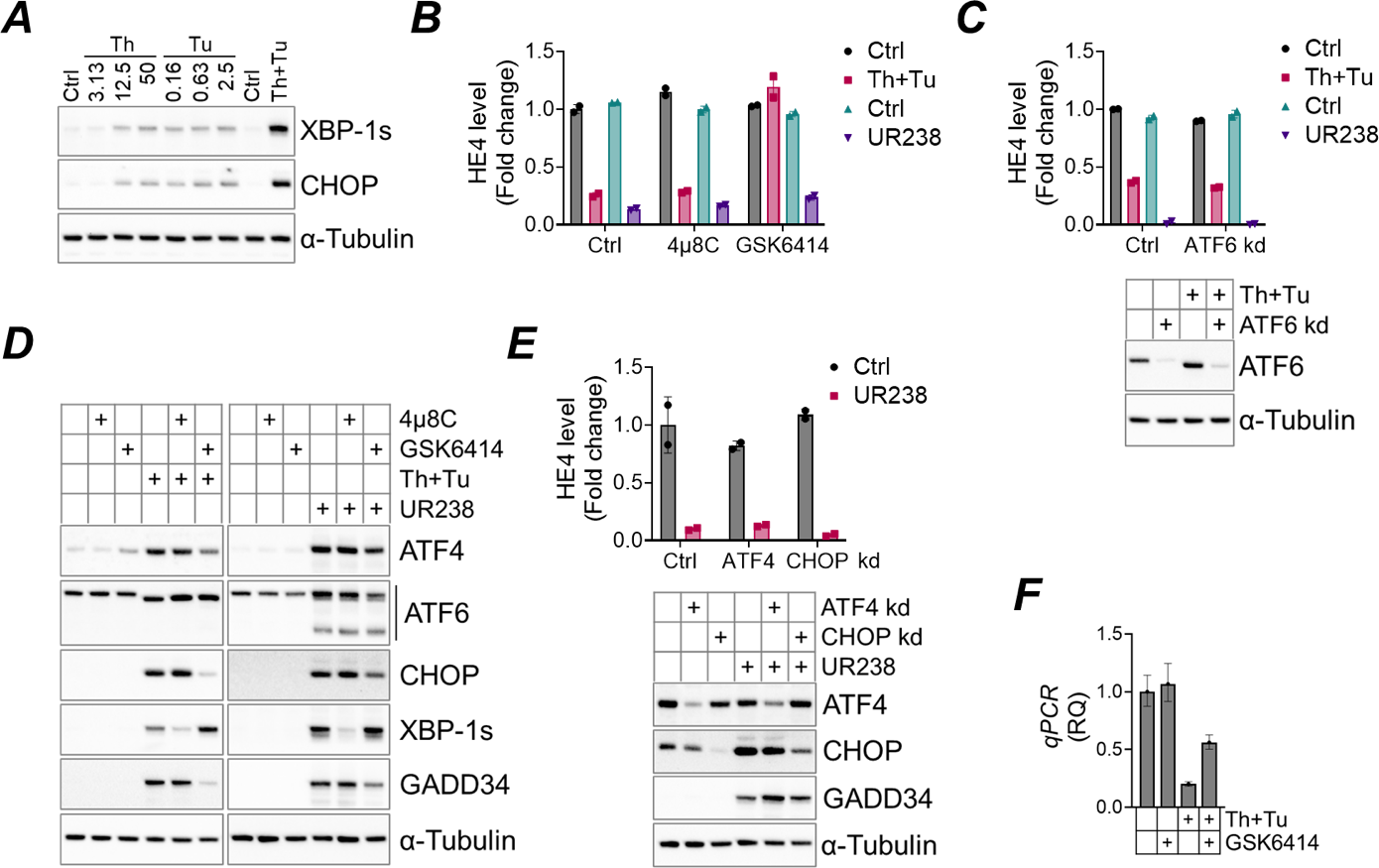
Figure 7. (A) ECC-1 cells were treated with thapsigargin (Th, nM), tunicamycin (Tu, μM), or both (Th+Tu, 50 nM and 2.5 μM each) for 18 hours. Protein extracts were analyzed for XBP-1s and CHOP, with α-tubulin as a loading control. (B) ECC-1 cells were pretreated with 4μ8C (10 μM), GSK2606414 (GSK6414, 1 μM), or control for 1 hour, then treated with Th+Tu (50 nM and 2.5 μM each) or UR238 (1000 nM) for 24 hours. HE4 levels were measured by a HE4 ELISA as described in “Materials and Methods”. (C) As in (B), but cells were transfected with ATF6 siRNA for 24 hours prior to Th+Tu or UR238 treatment. (D) As in (B), but protein extracts were immunoblotted using antibodies against indicated target proteins. (E) ECC-1 cells were transfected with siRNAs against ATF4 or CHOP for 24 hours, then treated with UR238 (1000 nM) for another 24 hours. HE4 levels or other targets were analyzed by ELISA and immunoblotting, respectively. (F) ECC-1 cells were treated with Th+Tu (50 nM and 2.5 μM each), with or without GSK2606414 (1 μM) for 24 hours. Cells were lysed, and isolated RNAs were analyzed for HE4 via qRT-PCR using YWHAZ as reference
Discussion
It is projected that 120,000 individuals will be diagnosed with EC in the year 2050 which is almost double the number of cases diagnosed in 2015. Approximately 30% of these patients are expected to present with advanced-stage disease requiring systemic chemotherapy [3]. Although molecular testing has shown potential for personalized EC treatment, options are still limited for patients who progress after standard care therapies and/or are ineligible for targeted therapies [4-7]. Further research is needed to identify newer drug-responsive targets and to develop new therapies for these patients. Prior research has demonstrated total 20S proteasome activity is higher in EC tumor tissue compared to intact endometrium, indicating that it is a protein exploited by cancer cells; therefore, proteasome inhibition may be a viable therapeutic option for advanced EC [9]. Though no proteosome inhibitors are yet approved in solid tumors, there have been some promising early phase clinic studies in gynecologic cancers which utilized protostome inhibitors in combination with standard chemotherapies [40].
FDA approved proteasome inhibitors come in two different classes: boronic acids (bortezominb and ixazomib) and epoxymycin derivatives (carfilzomib). Since, boronic acids (bortezomib) exhibit poor pharmacodynamics against solid tumors, we surmised that epoxymycin derivative carfilzomib could exhibit durable control over EC if improvement in the CLogP value (> 5.8) could be achieved. Lower CLogP values improve a drugs bioavailability and therapeutic efficacy [14]. UR238 was designed to improve the medicinal chemistry including CLogP. UR238's CLogP value of 5 is superior when compared to currently clinically available proteasome inhibitors. In the current experiments we were able to demonstrate that that UR238 is superior in suppressing cancer cell proliferation/viability and inducing cell death compared to carfilzomib. Additionally, we demonstrated that, as expected, UR238 selectively inhibits chymotrypsin-like proteasome activity and subsequently activates UPR signaling. In vivo efficacy of UR238 was also confirmed as it significantly blocked the tumor growth of AN3CA xenografts in NSG mice.
Exploiting ER stress to achieve cellular apoptosis also appears to be a promising therapeutic target in the treatment of cancer. In tumor cells which are continuously dividing and challenged by conditions such as restricted nutrients, DNA damage, metabolic stress and hypoxia, an upregulated UPR may be adaptive in maintaining their growth [41]. Studies have previously demonstrated increased ER stress markers in multiple cancer types including breast, prostate, pancreatic cancer, lymphoma, brain cancer, and colorectal cancer [42-52]. Additionally, in many cancers, high BiP levels have been found to correlate with higher pathological grade and aggressive phenotypes [53-55]. Analyzing uterine cancer proteomic datasets further support these conclusions as demonstrated by increased BiP, ATF6 and PERK protein abundance in tumors compared to adjacent normal tissues (Figure 6A). In EC specifically, Cali et al previously showed the attenuation of BiP resulted in a decreased growth rate and reduced invasion rate of EC cells, suggesting that UPR signaling not only plays a role in tumor growth but also cell migration and invasiveness [56]. Because EC cells, particularly those from more advanced disease, exhibit elevated levels of ER stress, they may be more susceptible to therapies that further disrupt ER homeostasis. This vulnerability offers a potential therapeutic window for selective targeting and may provide a novel treatment option for patients suffering from metastatic and advanced disease where treatment options are currently limited.
A notable feature of 20S inhibition by UR238 is the down regulation of HE4 expression. Of note, UR238 appears to regulate HE4 independently of PERK in contrast to tunicamycin and thapsigargin. In terms of clinical application of UR238, serum HE4 ELISA assay could serve as a surrogate marker for UR238 response in patients. HE4 is correlated with poor prognostic factors in patients with EC (stage, myometrial invasion and lymph node metastases) which suggest its use in monitoring treatment response and/or disease relapse [57]. We may find that treating with UR238 restores chemotherapy sensitivity through its impact on HE4.
Conclusion
In conclusion, we have shown for the first time that EC is dependent on proper function of the 20S proteasome machinery, and that proteasomal inhibition with UR238 blocks EC proliferation both in vitro and in vivo significantly. Though there are proteasome inhibitors already in clinical use, our 3rd generation proteasome inhibitor, UR238, is more effective than Carfilzomib in EC cells. UR238 exhibits longer duration of ubiquitination in EC cells, which can lead to sustained activities and reduced doses and hence increased safety indices compared to carfilzomib. Our study contributes to the body of evidence generated though our lab that UR238 is an active proteasome inhibitor with improved medicinal chemistry properties and a viable candidate 3rd generation proteasome inhibitor designed for the use in solid tumors. These experiments will support the advancement of UR238 for future pharmacokinetic/pharmacodynamic studies and the initiation of an investigational new drug application to test UR238 in phase-0/1 clinical trials. Utilizing UR238 for the treatment of EC would introduce a novel therapeutic pathway and option for the treatment of patients with endometrial cancer and other solid tumors.
Author contributions
MEW: Immunoblotting, cell viability experiments, data analysis, manuscript writing; KKK: Conceptualization, immunoblotting, experiments, data analysis, manuscript assembly; RKS: Animal experiments and design of UR238, synthesis and manuscript editing; AJM: Manuscript editing; NK: Immunoblotting; MEB: Immunoblotting; MSS: Data analysis; JPM: Cell culture; CWAS: Cell culture, data analysis; RWT: Editing, data review; RGM: Data review, editing, supervision and provision of resources.
Funding sources
MEW and KKK acknowledge Mae Goode Foundation for partially funding the studies presented in this study.
Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this manuscript.
Ethics statement
The animal studies conducted in this manuscript were performed with prior approval from University Committee on Animal Resources (UCAR) of the University of Rochester (Approval number: 2016-016E).
References
- Wijayabahu AT, Shiels MS, Arend RC, Clarke MA (2024) Uterine cancer incidence trends and 5-year relative survival by race/ethnicity and histology among women under 50 years. Am J Obstet Gynecol 231: 526.e1-526.e22. [Crossref]
- Surveillance E, End Results (SEER) National Cancer Institute 2025.
- American Cancer Society, Atlanta; 2026.
- Mirza MR, Chase DM, Slomovitz BM, dePont Christensen R, Novák Z, et al. (2023) Dostarlimab for primary advanced or recurrent endometrial cancer. N Engl J Med 388: 2145-2158. [Crossref]
- Eskander RN, Sill MW, Beffa L, Moore RG, Hope JM, et al. (2023) Pembrolizumab plus chemotherapy in advanced endometrial cancer. N Engl J Med 388: 2159-2170. [Crossref]
- Fader AN, Roque DM, Siegel E, Buza N, Hui P, et al. (2018) Randomized phase II trial of carboplatin-paclitaxel versus carboplatin-paclitaxel-trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. J Clin Oncol 36: 2044-2051. [Crossref]
- Meric-Bernstam F, Makker V, Oaknin A, Oh DY, Banerjee S, et al. (2024) Efficacy and safety of trastuzumab deruxtecan in patients with HER2-expressing solid tumors: Primary results from the DESTINY-PanTumor02 phase II trial. J Clin Oncol 42: 47-58. [Crossref]
- Brooks RA, Fleming GF, Lastra RR, Lee NK, Moroney JW, et al. (2019) Current recommendations and recent progress in endometrial cancer. CA Cancer J Clin 69: 258-279. [Crossref]
- Spirina LV, Kondakova IV, Koval’ VD, Kolomiets LA, Chernyshova AL, et al. (2012) Proteasome activity and their subunit composition in endometrial cancer tissue: Correlations with clinical morphological parameters. Bull Exp Biol Med 153: 501-504. [Crossref]
- Dimopoulos MA, Goldschmidt H, Niesvizky R, Joshua D, Chng WJ, et al. (2017) Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol 18: 1327-1337. [Crossref]
- Nooka AK, Kastritis E, Dimopoulos MA, Lonial S (2015) Treatment options for relapsed and refractory multiple myeloma. Blood 125: 3085-3099. [Crossref]
- Huang Z, Wu Y, Zhou X, Xu J, Zhu W, et al. (2014) Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol 10: 1795-1807. [Crossref]
- Manasanch EE, Orlowski RZ (2017) Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 14: 417-433. [Crossref]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23: 3-25. [Crossref]
- Bifulco G, Miele C, Di Jeso B, Beguinot F, Nappi C, et al. (2012) Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol Oncol 125: 220-225. [Crossref]
- Gray MJ, Mhawech‐Fauceglia P, Yoo E, Yang W, Wu E, et al. (2013) AKT inhibition mitigates GRP78 (glucose‐regulated protein) expression and contribution to chemoresistance in endometrial cancers. Int J Cancer 133: 21-30. [Crossref]
- Lu Q, Chen H, Senkowski C, Wang J, Wang X, et al. (2016) Recombinant HE4 protein promotes proliferation of pancreatic and endometrial cancer cell lines. Oncol Rep 35: 163-170. [Crossref]
- Li J, Chen H, Mariani A, Chen D, Klatt E, et al. (2013) HE4 (WFDC2) promotes tumor growth in endometrial cancer cell lines. Int J Mol Sci 14: 6026-6043. [Crossref]
- Rowswell-Turner RB, Singh RK, Urh A, Yano N, Kim KK, et al. (2021) HE4 overexpression by ovarian cancer promotes a suppressive tumor immune microenvironment and enhanced tumor and macrophage PD-L1 expression. J Immunol 206: 2478-2488. [Crossref]
- Kim K, Khazan N, Rowswell-Turner RB, Singh RK, Moore T, et al. (2024) Forchlorfenuron-induced mitochondrial respiration inhibition and metabolic shifts in endometrial cancer. Cancers 16: 976. [Crossref]
- Kim K, Khazan N, McDowell JL, Snyder CW, Miller JP, et al. (2024) The NF-κB-HE4 axis: A novel regulator of HE4 secretion in ovarian cancer. PLoS One 19: e0314564. [Crossref]
- Vichai V, Kirtikara K (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 1: 1112-1116. [Crossref]
- Khan RZ, Badros A (2012) Role of carfilzomib in the treatment of multiple myeloma. Expert Rev Hematol 5: 361-372. [Crossref]
- Darling NJ, Cook SJ (2014) The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta 1843: 2150-2163. [Crossref]
- Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ 22: 377-388. [Crossref]
- An WG, Hwang SG, Trepel JB, Blagosklonny MV (2000) Protease inhibitor-induced apoptosis: Accumulation of wt p53, p21WAF1/CIP1, and induction of apoptosis are independent markers of proteasome inhibition. Leukemia 14: 1276-1283. [Crossref]
- Drexler HC, Pebler S (2003) Inducible p27Kip1 expression inhibits proliferation of K562 cells and protects against apoptosis induction by proteasome inhibitors. Cell Death Differ 10: 290-301. [Crossref]
- Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: Cell life and death decisions. J Clin Invest 115: 2656-2664. [Crossref]
- Song S, Tan J, Miao Y, Li M, Zhang Q (2017) Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol 232: 2977-2984. [Crossref]
- Yoshii SR, Mizushima N (2017) Monitoring and measuring autophagy. Int J Mol Sci 18: 1865. [Crossref]
- Gammoh N, Lam D, Puente C, Ganley I, Marks PA, et al. (2012) Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci 109: 6561-6565. [Crossref]
- Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, et al. (2011) Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro Oncol 14: 215-221. [Crossref]
- Podar K, Gouill SL, Zhang J, Opferman JT, Zorn E, et al. (2008) A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene 27: 721-731. [Crossref]
- Hu H, Tian M, Ding C, Yu S (2019) The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol 9: 3083. [Crossref]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, et al. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18: 3066-3077. [Crossref]
- Wang D, Qian X, Du YC, Sanchez-Solana B, Chen K, et al. (2023) cProSite: A web based interactive platform for online proteomics, phosphoproteomics, and genomics data analysis. J Biotechnol Biomed 6: 573. [Crossref]
- Moore RG, Brown AK, Miller MC, Badgwell D, Lu Z, et al. (2008) Utility of a novel serum tumor biomarker HE4 in patients with endometrioid adenocarcinoma of the uterus. Gynecol Oncol 110: 196-201. [Crossref]
- James NE, Chichester C, Ribeiro JR (2018) Beyond the biomarker: Understanding the diverse roles of human epididymis protein 4 in the pathogenesis of epithelial ovarian cancer. Front Oncol 8: 124. [Crossref]
- Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69: 169-181. [Crossref]
- Ramirez PT, Landen Jr CN, Coleman RL, Milam MR, Levenback C, et al. (2008) Phase I trial of the proteasome inhibitor bortezomib in combination with carboplatin in patients with platinum-and taxane-resistant ovarian cancer. Gynecol Oncol 108: 68-71. [Crossref]
- Ojha R, Amaravadi RK (2017) Targeting the unfolded protein response in cancer. Pharmacol Res 120: 258-266. [Crossref]
- Lee E, Nichols P, Spicer D, Groshen S, Yu MC, et al. (2006) GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res 66: 7849-7853. [Crossref]
- Déry MA, Jodoin J, Ursini-Siegel J, Aleynikova O, Ferrario C, et al. (2013) Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res 15: R22. [Crossref]
- Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, et al. (2013) Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res 15: R2. [Crossref]
- Pike LR, Singleton DC, Buffa F, Abramczyk O, Phadwal K, et al. (2013) Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J 449: 389-400. [Crossref]
- Davies MP, Barraclough DL, Stewart C, Joyce KA, Eccles RM, et al. (2008) Expression and splicing of the unfolded protein response gene XBP‐1 are significantly associated with clinical outcome of endocrine‐treated breast cancer. Int J Cancer 123: 85-88. [Crossref]
- Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, et al. (2013) Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI (3, 4, 5) P3 production. PloS One 8: e80071. [Crossref]
- Gupta S, McGrath B, Cavener DR (2009) PERK regulates the proliferation and development of insulin-secreting beta-cell tumors in the endocrine pancreas of mice. PloS One 4: e8008. [Crossref]
- Su R, Li Z, Li H, Song H, Bao C, et al. (2010) Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer 10: 20. [Crossref]
- Overley-Adamson B, Artlett CM, Stephens C, Sassi-Gaha S, Weis RD, et al. (2014) Targeting the unfolded protein response, XBP1, and the NLRP3 inflammasome in fibrosis and cancer. Cancer Biol Ther 15: 452-462. [Crossref]
- Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, et al. (2008) GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol 10: 236-243. [Crossref]
- Li Z, Zhang L, Zhao Y, Li H, Xiao H, et al. (2013) Cell-surface GRP78 facilitates colorectal cancer cell migration and invasion. Int J Biochem Cell Biol 45: 987-994. [Crossref]
- Fu R, Yang P, Wu HL, Li ZW, Li ZY (2014) GRP78 secreted by colon cancer cells facilitates cell proliferation via PI3K/Akt signaling. Asian Pac J Cancer Prev 15: 7245-7249. [Crossref]
- Kaira K, Toyoda M, Shimizu A, Imai H, Sakakura K, et al. (2016) Decreasing expression of glucose‐regulated protein GRP78/BiP as a significant prognostic predictor in patients with advanced laryngeal squamous cell carcinoma. Head Neck 38: 1539-1544. [Crossref]
- Matsuo K, Gray MJ, Yang DY, Srivastava SA, Tripathi PB, et al. (2013) The endoplasmic reticulum stress marker, glucose-regulated protein-78 (GRP78) in visceral adipocytes predicts endometrial cancer progression and patient survival. Gynecol Oncol 128: 552-559. [Crossref]
- Calì G, Insabato L, Conza D, Bifulco G, Parrillo L, et al. (2014) GRP78 mediates cell growth and invasiveness in endometrial cancer. J Cell Physiol 229: 1417-1426. [Crossref]
- Behrouzi R, Barr CE, Crosbie EJ (2021) HE4 as a Biomarker for Endometrial Cancer. Cancers 13: 4764. [Crossref]