Introduction
Granulomatosis with Polyangiitis (GPA) is the most common Anti-Neutrophil Cytoplasmic Antibody (ANCA) associated vasculitis (previously called Wegener’s Granulomatosis) and is a rare multisystem autoimmune disease categorized by a combination of upper airway disease, lower airway disease, and glomerulonephritis [1]. About 11 million cases arise every year, equally distributed amongst males and females [2]. Multiple large prospective studies have found over 90% of those affected by the disease to be Caucasian, while only 1-4% identify as African American, Hispanic or Asian [3]. Patients typically present in the 4th or 5th decade of life with various nonspecific symptoms including hemoptysis, stridor, hoarseness and wheezing [4]. Systemic symptoms such as fever, weight loss, myalgias and headache can also be present, usually for several weeks to months [5]. This can make ANCA vasculitis difficult to diagnose as these presentations often overlap with other pulmonary diseases, such as malignancy [4]. Studies have found that prompt recognition and intervention is the most important factor in the morbidity and mortality of these patients [6]. Therefore, early detection (and hence, early treatment) is critical, although it may be difficult if another diagnosis is perceived as more likely on presentation. Clinicians need to consider ANCA associated vasculitides when patients present with pulmonary lesions in order to avoid delay in treatment of this condition [7]. This is especially true in the case of GPA, where early treatment leads to clinical remission in up to 75% of patients, but a rapidly progressive disease process and high rate of mortality may occur if diagnosis is delayed [4, 8]. We present a case of a patient who presented with weight loss, intermittent cough, an extensive smoking history and cavitating lung nodules. He underwent several biopsies with a high index of suspicion for malignancy and was not diagnosed with GPA until he presented with rapidly progressive glomerulonephritis 2 months later (Figure 1). Therefore we recommend that GPA should be included in the differential diagnosis of cavitary lung lesions and clinicians should always consider sending a urinalysis (UA), a cheap and easily administered test, in cavitary lung disease.
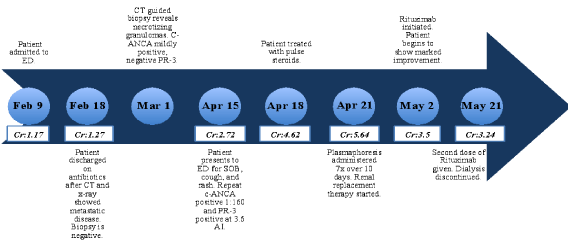
Figure 1: Timeline charting patient's clinical course.
Case Study
A 66 year old male with a 40 pack year smoking history presented to our hospital for fever and cough for 2 months. Family history was unknown as the patient was adopted. In the emergency department (ED) he had a fever of 38ºС and chest x-ray showed right upper lobe consolidation. Chest Computed Tomography (CT) revealed 8.3 x 9.4 cm cavitating right upper lobe lesion extending to the right hilum, bilateral hilar and mediastinal adenopathy and innumerable, bilateral parenchymal and pleural-based nodules (Figure 2). The patient was admitted with a diagnosis of pneumonia with a high suspicion for stage 4 lung cancer. He was treated with antibiotics and underwent endobronchial biopsy, transbronchial needle aspiration, and brushing and washing of the right upper lobe lesion. These samples were negative for malignancy, granulomas and acid fast bacilli. He was discharged on amoxicillin-clavulanic acid. As an outpatient he underwent a CT-guided biopsy of the right lower lobe lesion. Pathology demonstrated necrotizing granulomas, negative for acid fast bacilli. An ANCA panel was ordered which revealed a mildly positive c-ANCA (1:40), but a negative anti-Proteinase-3 (anti-PR3).
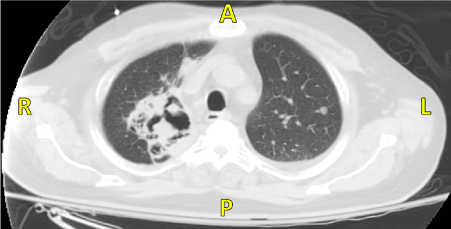
Figure 2: CT revealing 8.3 x 9.4 cm cavitating right upper lobe lesion extending to the right hilum with bilateral hilar and mediastinal adenopathy.
2 weeks later, the patient was admitted to the hospital with ongoing shortness of breath. A metabolic profile demonstrated a creatinine of 2.72 mg/dl and a UA with 3+ blood and trace protein. Repeat c-ANCA was positive at 1:160, but anti-PR3 was positive at 3.6 Antibody Index (AI). CT revealed no change in the dominant cavitating right upper lobe mass or in the suspected bilateral pulmonary metastases. Over the next few days his renal function worsened and creatinine increased to 5.68 mg/dl on hospital day 3. Renal biopsy performed that day revealed segmental necrotizing and crescentic glomerulonephritis with granulomatous features, severe interstitial inflammation with multifocal necrotizing interstitial granulomas and arteriolitis (Figures 3 a,b).
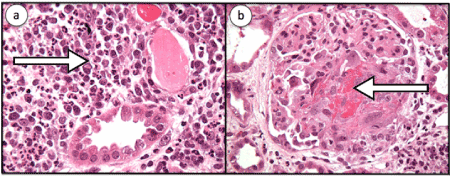
Figure 3: a. Renal biopsy was performed on that day which revealed severe interstitial inflammation (shown by the arrow), severe, with multifocal necrotizing interstitial granulomas and arteriolitis as well as b. Segmental necrotizing and crescentic glomerulonephritis (shown by the arrow) with granulomatous features. Granulomatous inflammation is a rare feature in renal pathology of GPA.
The patient was treated with pulse steroids for 3 days (1 gram of methylprednisolone per day), followed by plasmapheresis (7 doses total over 10 days with albumin replacement) and prednisone (60 mg per day). Due to progressive azotemia and volume overload, renal replacement therapy was initiated during this time as well. Although cyclophosphamide was considered for treatment, he had severe pancytopenia, so rituximab (1000 mg) was administered following completion of plasmapheresis. The patient’s pulmonary symptoms improved dramatically with steroid therapy (Figure 4). He was discharged to the hospital’s rehabilitation service 4 weeks after admission. While on this service, his creatinine stabilized at 3.2 mg/dl, so dialysis was discontinued. A second dose of rituximab was scheduled upon discharge.
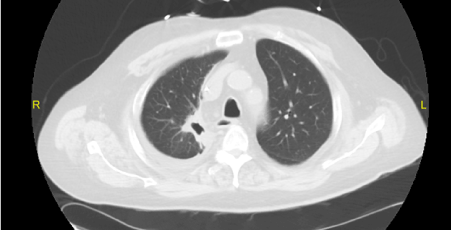
Figure 4: CT performed after treatment with rituximab with significant improvement of the right upper lobe cavitary lesion
Discussion
Diagnosis of Systemic Vasculitis
Historically, diagnosis of systemic vasculitides has been challenging as diagnostic criteria are constantly evolving and pathogenesis is not well understood. Factors including patient age, gender, ethnicity, affected vessel size, and the presence of granulomatous inflammation or specific antibodies must be considered as many disease processes may overlap [9]. In 1990 the American College of Rheumatology formulated a definition of GPA for nomenclature purposes to include necrotizing granulomatous inflammation within the upper or lower respiratory tract, vasculitis within small to medium sized blood vessels, and commonly associated simultaneous glomerulonephritis (Figure 5) [10]. In 2012 the Chapel Hill Consensus Conference expanded on this definition to include ANCA antibodies [11]. However, the intention of this meeting was not meant for clinical application but instead for research purposes. To date, no diagnostic criteria for GPA exists [12]. The development of clinical guidelines is important as our patients anti-PR3 was initially negative and universal clinical guidelines may prevent missed opportunities for diagnosis in the future.
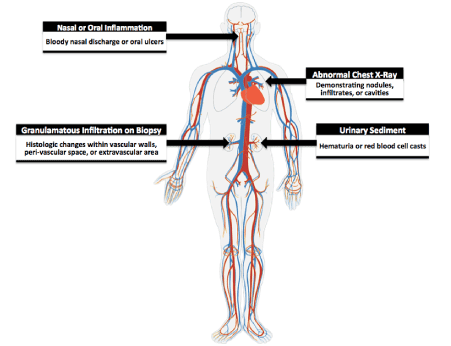
Figure 5: Criteria for GPA. The American College of Rheumatology classifies a patient to have GPA if they present with at least 2 of these 4 symptoms (sensitivity is 88.2% and specificity 92.0%).
The specificity and sensitivity of serologic testing in GPA is directly related to the extent and activity of the disease [4]. In the generalized form involving several organ systems, the specificity of the c-ANCA test is over 90% [12,13]. Amongst these patients, over 90% will be positive for anti-PR3 ANCA. In limited disease with only pulmonary or renal involvement, up to 40% of ANCA may be negative [14]. Although our patient’s symptoms reflected an active inflammatory disease process, anti-PR3 was surprisingly negative at the time of presentation. The presence of abnormal sediment, can indicate a significant amount of inflammation. This finding can reflect dysfunction in various areas of the kidney including the tubules, vasculature, or the interstitium [15]. Therefore an abnormal urine sediment, when another diagnosis is less likely, should lead the clinician to consider renal biopsy as was the case in our patient.
ANCA Panels
ANCA associated vasculitides may be present in several diseases other than GPA, such as microscopic Polyangiitis and Eosinophilic granulomatosis with Polyangiitis. ANCA panels can confirm the presence of an ANCA associated vasculitis and reveal a distinct diagnosis. Enzyme linked immunosorbent assay (ELISA) and staining by indirect immunofluorescence (IIF) are used to detect anti-PR3 and MPO ANCA [16]. ELISA is the newer method and its advantages include the speed at which one can obtain results, ease of performance, cost, efficiency and portability [26]. The disadvantages of ELISA include low sensitivity, especially in regards to p-ANCA related disease [17]. By combining ELISA and IIF, both a high specificity (98%) and sensitivity (75%) can be reached. Studies suggest that an initial screening of the patient using ELISA, followed by a confirmatory IIF test maximizes the efficiency of these tests [18].
Pathogenesis
Further understanding of disease pathogenesis may one day enable the advancement of screening and future treatment modalities. Although not well understood, GPA is a complex and multifactorial disease process influenced by factors including genetics, environment and autoimmunity. The relative risk of developing vasculitis in first degree relatives of a patient with GPA is 1.56 and genes regulating immunologic response, including Major Histocompatibility Complex and Proteinase 3 (which codes for anti-PR3), have been identified to be altered in individuals with GPA [19,20]. Environmental factors including occupation and infection have been associated with an increased risk of developing symptomatic and progressive disease [21].
In animal models, anti-PR3 has been identified as a component within cytoplasmic granules of neutrophils. In GPA, neutrophils may be activated by outlying agents such as cytokines, tumor necrosis factor, and the Fc portion of IgG4 antibodies to catalyze a respiratory burst. This process triggers a cascade of reactions which results in neutrophil aggregation with monocytes and T-cells, resulting in the formation of granulomas [14]. Recently, Lysosomal Membrane Protein 2 (LAMP-2) antibodies have been identified to represent a subtype of ANCA antibodies and may be important in the pathogenesis of ANCA associated vasculitis. LAMP-2 is a glycosylated membrane protein which is located on the membranes of anti-PR3 containing intracellular vesicles, as well as neutrophil outer membranes and endothelium [21]. Kain et al found that LAMP-2 has 100% homology with a portion of bacterial fimbriae, FimH, which may play a key role in endothelial destruction following infection [22].
Treatment
Different treatment regimens for GPA are currently under exploration, with the combination of cyclophosphamide and glucocorticoids as the current standard of care and effective in 70-90% of patients [23,24]. Cyclophosphamide has several adverse reactions including increased susceptibility to infection, bone marrow toxicity and predisposition to future malignancies [24]. Recent trials with rituximab have shown a reduction in adverse events, yet no significant difference in mortality [24,25]. Hence, for many patients with systemic vasculitis, rituximab has become preferable to cyclophosphamide for therapy. Plasmapheresis was added to our patient's regimen to quickly decrease the load of pathogenic ANCA. The combination of cyclophosphamide and plasmapheresis has been utilized in patients with severe renal failure, with studies suggesting a reduction from 60% to 40% of patients needing dialysis 1 year following treatment [26]. However, no existing large multi-modal trials study the combination of rituximab with plasmapheresis indicating that future studies may be warranted in order to optimize care.
What Makes Our Case Unique
The typical differential diagnosis for cavitating lesion is broad and may include infection, malignancy, ischemic changes, or dilation and displacement of lung tissue [27]. Our case illustrates the challenge a clinician may face when trying to make a diagnosis of GPA in a patient who presents atypically. Studies have shown that lung nodules in GPA rarely cavitate unless they are larger than 4 cm [28]. In our patient, the nodule was only 2.7 cm in diameter. The most common pulmonary manifestations of GPA include pulmonary nodules and airspace opacities. Cavitation of nodules is seen in about half the cases and are usually bilateral. Our patient presented with cavitating, unilateral lesions.
2021 Copyright OAT. All rights reserv
Over 70% of patients develop renal involvement during the course of the disease [29]. Generally, renal symptoms can take up to 2 years to develop [4]. Our patient’s disease progression and need for dialysis occurred within 2 months. Early identification and identification is crucial, as ANCA activation of neutrophils leads to oxidative damage to endothelial cells and patients develop a crescentic and destructive form of glomerulonephritis. As a result, when patients progress to advanced renal failure the prognosis becomes dire: 50% of patients will not regain renal function in the year following diagnosis.
Our patient's initial presentation reflects the typical lung involvement associated with GPA, however several factors including negative anti-PR3 in combination with an extensive smoking history resulted in delay in correct diagnosis and rapidly progressing disease. We have presented a case of a patient whose history of smoking, initially negative anti-PR3 and atypical imaging resulted in a delay in diagnosis. It is therefore important for clinicians to be aware of these variants in order to prevent delay in accurate diagnosis and poor patient outcomes. This is especially true when a cost-effective and easily accessible screening method such as a UA may be collected.
References
- 1 Pesci A, Manganelli P (2007) Respiratory system involvement in antineutrophil cytoplasmic-associated systemic vasculitides: clinical, pathological, radiological and therapeutic considerations. Drugs R D 8: 25-42.
- 2 Ntatsaki E, Carruthers D, Chakravarty K, D'Cruz D, Harper L, et al. (2014) BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology (Oxford) 53: 2306-2309. [Crossref]
- 3 Seo JB, Im JG, Chung JW, Song JW, Goo JM, et al. (2000) Pulmonary vasculitis: the spectrum of radiological findings. Br J Radiol 73: 1224-1231. [Crossref]
- 4 Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, et al. (1992) Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 116: 488-498. [Crossref]
- 5 Berden A, Göçeroglu A, Jayne D, Luqmani R, Rasmussen N, et al. (2012) Diagnosis and management of ANCA associated vasculitis. BMJ 344: e26. [Crossref]
- 6 Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM (1990) Pulmonary Wegener's granulomatosis. A clinical and imaging study of 77 cases. Chest 97: 906-912. [Crossref]
- 7 Homma S, Suzuki A, Sato K (2013) Pulmonary involvement in ANCA-associated vasculitis from the view of the pulmonologist. Clin Exp Nephrol 17: 667-671. [Crossref]
- 8 Rockall AG, Rickards D, Shaw PJ (2001) Imaging of the pulmonary manifestations of systemic disease. Postgrad Med J 77: 621-638. [Crossref]
- 9 Saleh A, Stone JH (2005) Classification and diagnostic criteria in systemic vasculitis. Best Pract Res Clin Rheumatol 19: 209-221. [Crossref]
- 10 Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, et al. (1990) The American College of Rheumatology criteria for the classification of Wegener's granulomatosis. Arthritis Rheum 33: 1101-1107. [Crossref]
- 11 Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, et al. (2013) 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65: 1-11. [Crossref]
- 12 Lutalo PM, D'Cruz DP2 (2014) Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener's granulomatosis). J Autoimmun 48-49: 94-8. [Crossref]
- 13 Katzenstein AL, Locke WK (1995) Solitary lung lesions in Wegener's granulomatosis. Pathologic findings and clinical significance in 25 cases. Am J Surg Pathol 19: 545-552. [Crossref]
- 14 Gomez-Puerta JA, Hernández-Rodríguez J, López-Soto A, Bosch X (2009) Antineutrophil cytoplasmic antibody-associated vasculitides and respiratory disease. Chest 136: 1101-1111. [Crossref]
- 15 Madaio MP (1990) Renal biopsy. Kidney Int 38: 529-543. [Crossref]
- 16 Wiik A (2002) Rational use of ANCA in the diagnosis of vasculitis. Rheumatology (Oxford) 41: 481-483. [Crossref]
- 17 Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, et al. (1998) Diagnostic value of standardized assays for antineutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. Kidney Int 53: 743-53. [Crossref]
- 18 Godbole MS, Valenzuela R, Deodhar SD, Calabrese L, Tubbs RR (1995) Comparative Study of ELISA and Indirect Immunofluorescence for the detection of Anti-Neutrophil Cytoplasmic Antibodies: Evaluation of the SCIMEDX/EURO Diagnostica ELISA assay in a Clinical Setting. American Journal of Clinical Pathology Dec 104: 667-672. [Crossref]
- 19 Knight A, Sandin S, Askling J (2008) Risks and relative risks of Wegener's granulomatosis among close relatives of patients with the disease. Arthritis Rheum 58: 302-307. [Crossref]
- 20 Willcocks LC, Lyons PA, Rees AJ, Smith KG (2010) The contribution of genetic variation and infection to the pathogenesis of ANCA-associated systemic vasculitis. Arthritis Res Ther 12: 202. [Crossref]
- 21 Duna GF, Cotch MF, Galperin C, Hoffman DB, Hoffman GS (1998) Wegener's granulomatosis: role of environmental exposures. Clin Exp Rheumatol 16: 669-674. [Crossref]
- 22 Kain R1, Exner M, Brandes R, Ziebermayr R, Cunningham D, et al. (2008) Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med 14: 1088-1096. [Crossref]
- 23 Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, et al. (2009) EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 68: 310-317. [Crossref]
- 24 Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, et al. (2010) Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363: 211-220. [Crossref]
- 25 Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, et al. (2010) Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 363: 221-232. [Crossref]
- 26 Luqmani RA (2014) State of the art in the treatment of systemic vasculitides. Front Immunol 5: 471. [Crossref]
- 27 Gadkowski LB, Stout JE (2008) Cavitary pulmonary disease. Clin Microbiol Rev 21: 305-333, table of contents. [Crossref]
- 28 Grotz, W, Mundinger A, Würtemberger G, Peter HH, Schollmeyer P (1994) Radiographic course of pulmonary manifestations in Wegener's granulomatosis under immunosuppressive therapy. Chest 105: 509-513. [Crossref]
- 29 Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, et al. (2007) Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 18: 2180-2188. [Crossref]