Abstract
This study was aimed to determine the frequency of idiopathic pulmonary fibrosis (IPF) diagnosis in different clinical services in the Instituto Nacional de Enfermedades Respiratorias “Dr. Ismael Cosio Villegas” (INER). Based on 2009-2013 INER biostatistics report, the frequency for IPF was determined. The files of patients with a HRCT compatible and probable to have usual interstitial pneumonia (UIP) were requested, diagnosis was confirmed and clinical, pulmonary function and treatment data of patients with confirmed diagnosis was analysed; the frequency of disease was re-calculated following compliance of criteria from Consensus 2011.
Diffuse interstitial lung diseases (ILD) accounted for 8.7% of admissions at INER, with 12.6% of them being diagnosed as IPF, which corresponds to 1.1% of total admissions at INER. Following Consensus 2011, 80 IPF cases were confirmed with the frequency reduced to 4.16% compared to ILD and to 0.36% compared to total admissions. This allowed us detect that there are great problems in diagnosis of IPF due to noncompliance of Consensus 2011 outside the clinical service for interstitial diseases.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a diffuse interstitial lung disease with a worst prognosis, with an average survival rate between 3 and 5 years following diagnosis, affecting primarily male population and subjects over 55 years of age [1]. Worldwide incidence is estimated to be 4.3 to 16.3 per 100,000 persons, with prevalence between 13 and 20 per 100,000 persons [2]. Its epidemiologic impact is unknown in Mexico. Its aetiology is unknown and clinically patients show cough without expectoration, dyspnoea, and progressive fatigue for at least 6-month evolution. Diagnose is established based on Consensus 2011 criteria (ATS/ERS/JRS/ ALAT) [1], where a pulmonary fibrosis secondary to other entities such as collagen-vascular diseases, pneumonitis due to hypersensitivity and work-related disease is recommended to be ruled out, additionally to confirmation of diagnosis by a high-resolution computerised tomography (HRCT) with a typical pattern for usual interstitial pneumonia or, if possible, obtaining a histological confirmation for usual interstitial pneumonia pattern by surgical lung biopsy. Also, the lung function should be assessed at diagnosis by spirometry, six minute walking distance test (6MWD), carbon monoxide diffusing capacity test (DLCO), and plethysmography. It is suggested to conduct a follow-up for disease evolution quarterly, by using respiratory function tests (spirometry, 6MWD, DLCO). Unfortunately, currently there is no a curative treatment; thus, it is important to obtain a correct diagnosis because the prognosis is more encouraging for the other ILD, with successful treatment schemes. In May, 2014 the results for two drugs (nintedanib and pirfenidone) were presented, with both delaying the disease progress and having IPF as the unique approved indication. This makes precise diagnosis even more important.
INER is a national referral site and educational hospital, providing clinical services for the most frequent pulmonary diseases such as lung cancer, tuberculosis, interstitial diseases, etc., and these services are provided by experienced, certified pulmonologists and training pulmonologists. Services are intended to admit patients for each according to their disease; however, it is not possible always, so any service may conduct a diagnostic study for the patient.
Therefore, the warrant for this study is that, although IPF diagnosis is made in the institute by pulmonologists, there is heterogeneity for accepting the use of diagnostic criteria in the different clinical services.
Methodology
A retrospective, observational and descriptive study was conducted. A request was made to INER Biostatistics Department for the total number of patients with a diagnosis of pneumology in adults at discharge for the period 2009-2013. They were reviewed and separated by pulmonary pathologies, determining the initial frequency of IPF; subsequently, the tomographic studies were reviewed by an experienced pulmonologist-radiologist and they underwent a consensus by the group in the clinic of interstitial diseases of INER, including a pathologist experienced in interstitial diseases and an experienced radiologist to determine the usual, possible or non-matching pattern. The clinical files for cases with a usual or probable pattern were reviewed to confirm diagnosis by using the Consensus 2011 criteria, the IPF frequency was determined and clinical variables were analysed.
All those patients reported with IPF diagnosis by the INER Biostatistics Department and with an IPF diagnosis at discharge from hospital for the 2009-2013 period were included in the study.
Frequencies, averages and standard deviation were used to analyse data for the different clinical, functional and treatment variables, by using Microsoft Excel version 14.4.7 software.
Results
Based on biostatistics report, we found that total number of diagnoses at discharge in adults for 209-2011 period at INER was 21,962 patients, with 1,923 of them having ILD (8.7% of total discharges), and 243 had an IPF diagnosis, accounting for 12.6% of ILD and 1.1% of total discharges (Figure 1). With respect to these 243 IPF reported cases, 47% (115 patients) occurred in Mexico City, 31% (76 patients) in the state of Morelos, and 13% (34 patients) in other states such as Puebla, Chiapas, Veracruz, Guerrero, Tlaxcala and Hidalgo. We don’t know if these patients were referred by private physicians, public hospitals or if they arrived by themselves.
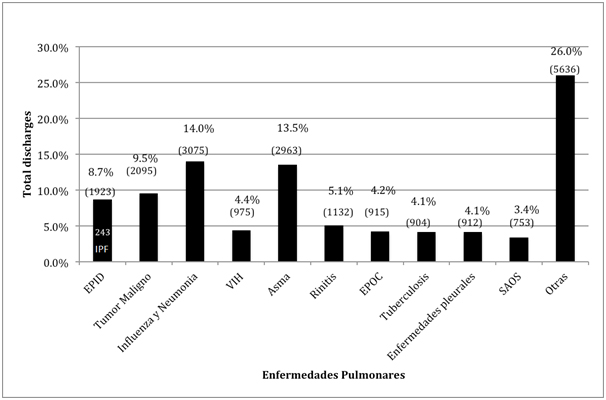
Figure 1. Diagnoses for discharges at INER for 2009-2013
Source: INER Biostatistics Report 2009-2013
Among 243 IPF initial cases, 92 were ruled out, 36 because they occurred in persons with less than 55 years of age, 25 because they had no HRCT available, and 31 because they were re-admissions, after which 151 cases remained, that based on high-resolution tomographic features were classified as a possible and non-matching usual interstitial pneumonia pattern according to Consensus 2011 criteria [1,2]. Non-matching cases were definitely excluded from study Figure 2.
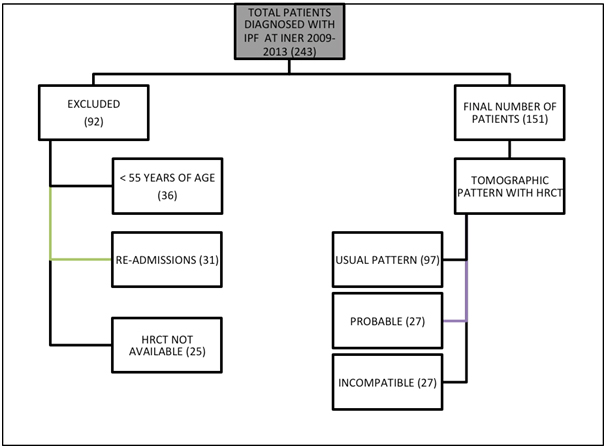
Figure 2. Initial classification of patients with IPF based on inclusion and exclusion criteria
Following tomographic classification, the files for 97 cases with usual interstitial pneumonia pattern and for 27 possible were reviewed, and we found that 6 of these 27 possible cases were confirmed by surgical lung biopsy and any other associated pathology was identified, thus prevailing 18 confirmed cases. Additionally, of the 97 cases with a usual interstitial pneumonia pattern, 35 were ruled out due to the presence of positive antibodies for any immune disease, with remaining 62 confirmed cases. Thus, at the end of sampling 80 confirmed cases prevailed (4.16% of total ILD and 0.36% of INER total discharges), 62 of which had a tomographic pattern compatible with usual interstitial pneumonia and 18 with possible usual interstitial pneumonia (Table 1).
Table 1. Number of cases having HRCT with usual or probable interstitial pneumonia pattern ruled out due to collagen-vascular disease or confirmed by lung biopsy
HRCT |
Number of cases |
Ruled out due to presence of immune disease |
Confirmed by Lung biopsy |
Final number of cases |
Final n |
Usual pattern |
97 |
35 |
18 |
62 |
80 |
Probable pattern |
27 |
9 |
6 |
18 |
Fuente: Biostatistics report for patients with IPF diagnosis at discharge |
Finally, the variables to be studied for the 80 confirmed cases were collected to analyse our variables, with the following results found: the occurrence of IPF is prevalent in male with a ratio 5:1 with respect to female patients [3-5]. The average age for the occurrence is 69 ± 8 years; the disease evolution lasts 24 ± 23 months from appearance of symptoms. Eighty percent of patients are ex-smokers. Associated comorbidities in frequency order are: systemic hypertension (30%), type 2 diabetes mellitus (T2D) (21%), chronic obstructive pulmonary disease (COPD) (16%), and obstructive sleep apnoea syndrome (OSA) (4%). These comorbidities were confirmed by relevant studies when files were reviewed; e.g., COPD was determined by spirometry and HRCT. Thirty percent of patients required a surgical pulmonary biopsy, whereas bronchoalveolar lavage (BAL) was conducted for 84% of cases (Table 2).
Table 2. Clinical characteristics of study population
Variable |
N=80 |
Gender: M:F |
68(85%):12(15%) |
Age (years)* |
69 (±8) |
Ex-smokers |
64 (80%) |
Current disease (months)* |
24 (±23) |
COPD |
13 (16%) |
OSA |
3 (4%) |
T2D |
17 (21%) |
SAH |
24 (30%) |
* Results are averages (SD)
Source: Files of patients with IPF reviewed by INER file service |
At diagnosis time, patients had a characteristic BAL with macrophage predominance of 84 ± 14%. Pulmonary function included a FVC of 68 ± 21%, FEV1 of 74 ± 21%, TLC of 62 ± 15%, DLCO of 52 ± 31%, PaO2 of 56 ± 12 mmHg, PaCO2 of 34 ± 5 mmHg, SaO2 at rest of 85 ± 12%, 6MW of 241 ± 156 m and PSAP of 45 ± 18 mmHg (Table 3).
Table 2. Clinical characteristics of study population
Variable |
N=80 |
Gender: M:F |
68(85%):12(15%) |
Age (years)* |
69 (±8) |
Ex-smokers |
64 (80%) |
Current disease (months)* |
24 (±23) |
COPD |
13 (16%) |
OSA |
3 (4%) |
T2D |
17 (21%) |
SAH |
24 (30%) |
* Results are averages (SD)
Source: Files of patients with IPF reviewed by INER file service |
Table 3. Characteristics of BAL, respiratory mechanics and pulmonary gas diffusion at diagnosis time
Variable |
Average (SD) |
Macrophage BAL |
84% (±14) |
Lymphocyte BAL |
13% (±10) |
Neutrophil BAL |
2% (±6) |
FVC |
2.05 L (±0.7) |
68% (±21) |
FEV1 |
1.80 L (±0.5) |
74% (±21) |
TLC |
62% (±15) |
DLCO |
52 (±31) |
PaO2 (mmHg) |
56 (±12) |
PCO2(mmHg) |
34 (±5) |
SaO2 |
85% (±12) |
Walk (m) |
241(±156) |
45 (±18) |
Finally, we found that there has been a substantial change in initial treatment of disease, as we analysed the treatment before and after 2011, when an alert occurred stating that triple scheme treatment (azathioprine, prednisone and N-acetylcysteine [NAC]) worsened the patients instead of benefit them [6]. We observed that before 2011 the treatment was based on the use of prednisone, followed by acetylcysteine, azathioprine, colchicine and finally pirfenidone (Figure 3), but following the above mentioned alert we can see that there has been a decrease in prescribing prednisone, azathioprine and colchicine, and an increase in the use of antifibrotics such as pirfenidone, which was approved by the Federal Commission for Protection against Sanitary Risks (COFEPRIS) for the use in Mexico. It is worthy to mention that during the conduct of this study NAC remained under use as final results from PANTHER study were yet unknown (Figure 4).
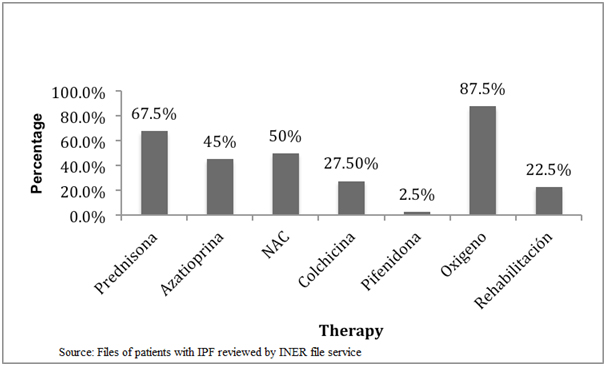
Figure 3. Initial therapy for IPF at INER from 2009 to 2011
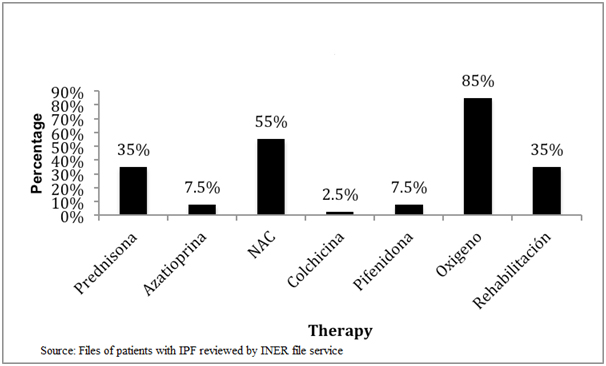
Figure 4. Initial therapy for IPF at INER from 2011 to 2013
Discussion
IPF is defined as a usual interstitial pneumonia with unknown cause, occurring with chronic and progressive pulmonary fibrosis, with a mean survival rate of 3-5 years following the diagnosis [7]. Its epidemiology is unknown in Mexico and other Latin America countries.
In our study, the frequency found at a national referral centre (INER) accounts for a 1.1% proportion of total admissions for pulmonary and airways pathologies, which means their frequency is low. Additionally, as in international guides [1], the predominance is higher in male than female patients, with men: women 5:1 ratio in our study and an average age of 69 years at diagnosis time (international guides report 66 years of age). A relation to smoking history was also apparent in 80% in our cohort, which is significant as, according to a study reported by Baumgarner et al. [8] as well as that conducted by Garcia-Sancho et al. [9], smoking status may play an important role as an IPF risk factor.
On the other hand, regarding comorbidities observed in patients with IPF, SAH occurred in 30% patients and T2D in 21%, which is not only impressive due to the frequency but also because of the metabolic disorder these diseases may cause alone, and even by COPD existing in up to 16% of IPF cases, leading to so called combined syndrome (IPF/pulmonary emphysema), which is reported at a frequency similar to that described by Mejia et al. in a previous study (28%) [10].
However, although IPF frequency is low, its consequence is fatal and this may be related to a very late diagnosis (an average of 2 years following starting symptoms, according to our results), which reveals that first contact physician does not suspect the diagnosis and, therefore, does not refer the patient early or the patient does not see a specialised physician. Due to delay for diagnosis, patients present with a severe depletion of respiratory function by the time of diagnosis, which is relevant because according to a study conducted by Steven et al. [11] in a clinic for interstitial diseases at Inova Fairfax Hospital, a FVC between 55 and 69% predicted classifies the patient as being at a moderate IPF stage where if DLco between 40 and 60% occurs, less than 40% patients will have a survival of 9 years following diagnosis.
Similarly, we can see that late diagnosis and subsequent decline in pulmonary function occur with complications at diagnosis time, reflected by the presence of pulmonary arterial hypertension (PAH), which is associated with up to 80% of all cases of tricuspid valve failure and subsequent progression to right valve failure. In a study, Harari et al. [12] observed that the only test with demonstrated significant differences between patients with and without PAH is the forced vital capacity (FVC) in spirometry, as 6MW test has been demonstrated to be different between both groups. However, we did not classify patients in our study as having IPF/PAH and IPF alone, so we did not compare pulmonary function tests. Once stated this, we can observe that based on our results, patients in our cohort presented IPF at moderate stage accompanied by mild PAH at diagnosis time, with expected survival of 9 years in less than 40%. Although Consensus 2011 does not include BAL as a necessary study for diagnosis, our group agrees the European society that BAL is a study that allows us rule out other type of interstitial disease and is a diagnostic tool requested by us to most of patients; for this study it was conducted for 84% of cases.
Currently, there are no curative treatments for IPF, but in recent studies it has been demonstrated that the use of antifibrotics drugs such as nintedanib or pirfenidone may significantly reduce the loss of FVC and increase tolerance to exercise compared to placebo group, with p=0.001 for both drugs, with a decrease of exacerbations favouring nintedanib [3,4].
With respect to this issue, in our study we can observe that drugs more frequently prescribed at INER are those included in the so called triple-therapy treatment (prednisone, azathioprine and acetylcysteine), which led to a significant decrease in prescribing, at least at INER, from 2011, when an alert was reported for PANTHER study [6], which makes sense with the theory developed by Dr. Selman [7] that IPF is not an inflammatory disease and neither an autoimmune-type disease, but rather a pulmonary epithelial disorder.
Finally, it should be emphasised that even though INER is a national referral centre, the compliance of Consensus 2011 criteria is unsatisfactory as our number of cases decreased from 243 initial cases to only 80 at the end of study, following the compliance of this consensus criteria, which means there is an over-diagnosis of 67.08% and only 32.92% present IPF; thus, continuous education and knowledge will be increased in all the clinical services, to improve diagnostic precision of interstitial diseases outside the service.
Conclusions
- Idiopathic pulmonary fibrosis has a low frequency with respect to total ILD diagnosed in our national referral centre for the period of study; however, we hope collect more reliable data once the national IPF register is implemented.
- In our experience, the poor prognosis for patients with IPF is related to: 1) a late diagnosis, 2) a decrease in pulmonary function due to disease progression, occurrence of complications and comorbidities, and 3) the lack of curative drugs for the disease.
- The non-compliance of diagnostic consensus for IPF leads to over-diagnosis and, consequently, to a wrong therapeutic approach with the corresponding medical and economic consequences.
- Existing disease over-diagnosis is high, so modifications to continuous medical education will be implemented and also with respect to a tight compliance of international criteria from Consensus 2011 of ATS/ERS/JRS/ALAT in the Institute.
References
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, et al. (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788-824. [Crossref]
- Ley B, Collard HR (2013) Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol 5: 483-492. [Crossref]
- King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, et al. (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370: 2083-2092. [Crossref]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, et al. (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071-2082. [Crossref]
- Xaubet A, Ancoechea J, Bollo E (2013) Normativa sobre el diagnostico y tratamiento de la fibrosis pulmonar idiopatica. Arch Bronchoneumol 49: 343-53.
- Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, et al. (2012) Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366: 1968-1977. [Crossref]
- Selman M, King TE, Pardo A, American Thoracic Society, European Respiratory Society, et al. (2001) Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 134: 136-151. [Crossref]
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA (1997) Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 155: 242-248. [Crossref]
- García-Sancho Figueroa MC, Carrillo G, Pérez-Padilla R, Fernández-Plata MR, Buendía-Roldán I, et al. (2010) Risk factors for idiopathic pulmonary fibrosis in a Mexican population. A case-control study. Respir Med 104: 305-309. [Crossref]
- Mejia M, Selman M (2009) Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 136: 10-5.
- Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, et al. (2011) Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest 140: 221-229. [Crossref]
- Harari S, Caminati A, Cassandro R, Conti S, Madotto F, et al. (2015) Pulmonary hypertension in idiopathic pulmonary fibrosis does not influence six-minute walk distance: results from a retrospective study. Sarcoidosis Vasc Diffuse Lung Dis 31: 297-305. [Crossref]