Abstract
This is a report of a female newborn baby who presented at the age of one hour after birth with signs of respiratory distress with marked cyanosis. The critical condition of the baby and the stormy course of the disorder highlighted the importance of starting immediate management and establishing an etiological diagnosis, as this will make the difference in the outcome and the prognosis.
Key words
cardiomyopathy, noncompaction, newborn baby
Clinical presentation
This was a female infant born at term after an uneventful pregnancy. Antenatal ultrasound did not show fetal anomaly and antenatal serology for infectious diseases was not significant. High vaginal swab from the mother was negative for group B streptococcus. Delivery was by elective Caesarean section because of previous section. Apgar Score was 9 and 10 at 1 and 5 minutes respectively. Birth weight was 2230 grams but the baby was vigorous with an oxygen saturation of 95% in room air at 15 minutes and was eventually sent to the normal newborn nursery.
At the age of one hour the pediatric doctor was called because the baby was showing signs of respiratory distress with marked cyanosis and an oxygen saturation ranging from 70% to 80% in room air. Clinical examination revealed no signs of dysmorphism but the baby was showing respiratory distress with marked cyanosis. There were no abnormal findings on examination of the cardiovascular system.
On admission to the neonatal intensive care unit (NICU) the baby was started on nasal continuous positive airway pressure (nCPAP) of 6 cm H2O, FiO2 of 0.3 but the oxygen saturation (SaO2) remained unchanged at 80% and below. Blood gases at that time showed a pH of 7.12, pCO2 of 45 mm Hg and a bicarbonate of 14 mmol/I with a base deficit of -13. With the increasing requirements of oxygen the baby was shifted from nCPAP to mechanical ventilation. In spite of reaching an inspiratory pressure of 26 cm H2O the baby remained hypoxemic at which point a diagnosis of persistent pulmonary hypertension (PPHN) was entertained and high frequency oscillatory ventilation (HFOV) was started together with inhaled nitric oxide. Soon after admission the baby was noticed to have systemic hypotension requiring inotropic therapy. Dopamine and dobutamine were titrated to a maximum of 20 micrograms per kilogram per minute after which epinephrine and hydrocortisone were instituted consecutively because of persistent hypotension. At this point the differential diagnosis rested between cyanotic congenital heart disease, septic shock or metabolic disorder and antibiotics were started.
On interviewing the family it was revealed that the parents were first-degree cousins with five living children but one of the siblings who was a male infant expired at the age of two days and the death was attributed to possible hypoxic ischemic encephalopathy, septic shock or a metabolic disorder most likely adrenal insufficiency.
Taking into account the clinical presentation and the family history the baby was investigated accordingly. A chest radiograph revealed normal lung fields with no cardiomegaly. Initial laboratory investigations for blood cell counts and C-reactive protein were not significant. Blood culture did not grow organism. Serum lactate and ammonia were within normal levels.
During cardiac monitoring of the baby it was noticed that the tracing was of low voltage and this was confirmed by electrocardiogram (ECG) (Figure 1).
2021 Copyright OAT. All rights reserv
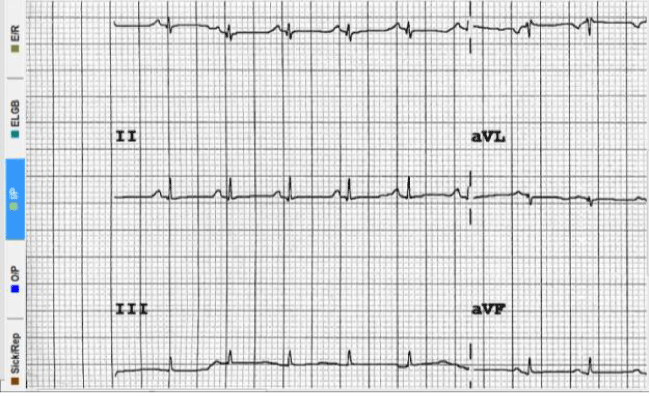
Figure 1. ECG showing low voltage
The ECG finding of low voltage is consistent with a myocardial insult. An immediate pediatric cardiology evaluation was sought and an echocardiogram confirmed the presence of a huge ductus arteriosus (PDA) with bidirectional flow and evidence of a cardiomyopathy in the form of noncompaction of the left ventricle with heart failure. The baby was started on furosemide and was continued on inotropic support. By the age of 24 hours the baby was still on HFOV, inhaled nitric oxide and maximum inotropic support plus epinephrine and hydrocortisone. Although the oxygenation improved and the blood gases normalized, the baby remained hypotensive. Repeat blood cell count and C-reactive protein did not show any significant changes. At the age of 48 hours the baby was still hypotensive but developed hypoglycemia (glucose 2.1 mmol/liter) and hyponatremia (serum sodium 126 mmol/liter). At this point we recalled that the previous sibling who expired had the same clinical course and findings that were suggestive of adrenal insufficiency and hence the dose of hydrocortisone was adjusted to the physiological one. The pediatric endocrine team stepped in and a full endocrine work up was carried out. Other investigations including creatinine kinase, liver function tests, Tandem MS, plasma amino acid screen and urine organic acids were all non-significant. The endocrine work up showed a non-elevated 17-hydroxyprogesterone and a high adrenocorticotrophic hormone (ACTH) at 303 picogram/ml (NR 7.2 -63.7), high renin, low cortisol and normal aldosterone. The ratio of plasma renin to aldosterone was 11:1. Studies for thyroid hormones revealed a thyroid stimulating hormone (TSH) of 0.3 mIU/liter (NR 0.5 – 4.5), free thyroxin (FT4) of 5.6 picomol/liter (NR 12- 22) and free tri-iodothyronine (T3) of 3.6 picomol/liter (NR 3- 6.8).
Taking all the results together it was obvious that we were dealing with a case of cardiomyopathy with noncompaction of the left ventricle [1], secondary hypothyroidism and primary adrenal insufficiency. Thyroxine was started and hydrocortisone therapy was continued. The baby continued to improve clinically and biochemically and ventilation and nitric oxide were weaned gradually with improvement in oxygenation. Hypotension started to improve and inotropes were titrated to lower doses. By the age of five days a repeat echocardiogram showed improvement of left ventricular function but there were still areas of noncompaction and the PDA became smaller. At the age of 13 days echocardiography revealed significant improvement in the myocardium and the left ventricular noncompaction and complete closure of the PDA. By then the baby was weaned totally from inotropes, oxygen and ventilation and continued treatment with thyroxin and hydrocortisone. Mineralocorticoid deficiency was ruled out because the ratio of plasma renin of 11:1 excludes that diagnosis. FT4 level returned to normal within one week but the ACTH level continued to rise before it settled after one month.
Further work up included imaging (MRI) of the pituitary, thyroid and adrenal glands which did not show abnormal findings.
Chromosomal karyotype was 46XX while whole exome sequence did not reveal clinically significant variants.
Echocardiography at the age of one month showed complete resolution of the noncompaction.
On follow up the baby is now ten month old with normal growth and development and is continued on thyroxin and hydrocortisone.
Discussion
This female infant has shown the classical echocardiographic findings of left ventricular noncompaction [2] (LVNCM), which we think is related to congenital thyroid hormone deficiency. LVNCM is a rare form of cardiomyopathy which has been described more recently in the literature. Most of the studies relating to the etiology of LVNCM come from experimental animal models and data from adult patients [3].
Studies of the embryology of the heart muscle show that during the first four weeks of fetal life, the myocardium is composed of meshwork of loosely interwoven muscle fibers of spongy appearance. This meshwork forms trabeculae alternating with recesses that communicate with ventricular cavity to provide blood supply to the cardiac muscle.
At 6 -8 weeks of fetal life the ventricular myocardium is gradually compacted and the recesses turn into capillaries. The process goes from the epicardium to the endocardium and from the heart base to the apex. Compaction is more complete in the left than the right ventricle. Noncompaction of the left ventricle represents persistence of multiple trabeculation in ventricular myocardium and is now considered as a form of cardiomyopathy.
Left ventricular noncompaction (LVNCM) is rare and is said to affect 0.14% of the pediatric population and has been reported in association with heterogeneous etiological conditions, which include a wide number of genetic disorders [3]. LVNCM can be sporadic or familial. The familial form can be inherited as autosomal recessive, autosomal dominant or sex-linked recessive. It has also been reported as an isolated disorder or in coexistence with structural heart diseases and can be complicated by congestive heart failure, arrhythmias and thromboembolic events.
A number of disorders have been reported in association with LVNCM including chromosomal trisomies, neuromuscular disorders, inborn errors of metabolism and a variety of other syndromes [3] The reported inborn errors of metabolism include Barth syndrome, malonyl co-enzyme A carboxylase deficiency and mitochondrial disorders [3] .
In our baby we had two endocrine defects namely secondary hypothyroidism and primary adrenal insufficiency. Hypothyroidism is clearly secondary because thyroid hormone (FT4) was low with a low thyroid stimulating hormone (TSH). Adrenal insufficiency was confirmed with very high adrenocorticotrophic hormone (ACTH), very high renin and low cortisol levels. A plasma renin to aldosterone ratio of 11:1 ruled out mineralocorticoid deficiency because the accepted diagnostic ratio is considered when it is above 30:1. Therefore the adrenal insufficiency here represents an isolated glucocorticoid deficiency because there was no evidence of androgen excess in our female baby. In this case we hypothesize that the LVNCM cardiomyopathy has resulted from the deficiency of the thyroid hormone, which is an important mediator in the cardiomyocyte development as is discussed below. The family history of a previous affected male sibling suggests an autosomal recessive inheritance in this case.
The involvement of the thyroid hormone in the cardiac myocyte development is well established4. The thyroid gland synthesizes tetra-iodothyronine (Thyroxin- T4) and tri-iodothyronine (T3) in response to thyroid stimulating hormone (TSH) secreted by the anterior pituitary. The secreted thyroxin makes 85% of the thyroid hormone and is biologically inactive. Thyroxin is converted to T3 by monodeiodination in the liver, kidney and skeletal muscle. The heart relies mainly on T3 activity because myocyte intracellular deiodinase activity in insignificant [4]. Thus T3, and not T4, is transported to the cardiac myocyte. In the myocyte T3 exerts its cellular action through binding to thyroid hormone nuclear receptors (TRs). TRs mediate the induction of transcription by binding to thyroid hormone response elements (TREs) in promoter regions of positively regulated genes. Therefore while bound to T3, TRs induce transcription. In the absence of T3, TRs repress transcription [4]. The thyroid hormone effects on the cardiac myocyte are intimately associated with cardiac function via regulation of the expression of key structural and regulatory genes. The myosin heavy chain genes encode the two contractile proteins of the thick filament in the cardiac myocyte. These two proteins are largely responsible for enhanced contractile function and diastolic relaxation in the heart [4]. Deiodination of T4 to T3 is catalyzed by three types of deiodinases; D1, D2 and D3 [5]. These oxido-reductases differ in their catalytic properties, tissue distribution and developmental expression. Deiodination of T4 to active T3 is catalyzed by both D1 and D2 with D2 having 1000 fold higher affinity for T4 than D1. Whereas D1 is primarily expressed in the liver and kidneys, D2 is present in other tissues including the human heart and muscle. D2 provides T3 in tissues where it is expressed including the heart. D3 is involved in deiodination of the inner tyrosyl ring of T4, which generates the inactive reverse T3 (rT3). Studies have shown that different deiodinase activities are highly regulated both during development and adult life. Tight regulation of tissue T3 is particularly critical during development. Adult plasma levels of T3 are not compatible with normal fetal development and high D3 activity in pregnant uterus and placenta protects the fetus from too much maternal thyroid hormone. Spatial and temporal patterns of expression of D2 and D3 appear to precisely regulate the T3-dependent aspects of cell proliferation and particularly cell differentiation [5]. These molecular data may explain the persistence of noncompaction in our baby suggesting that failure of activation of T3 or its inactivation by D3 has played the major role in failure of progression of noncompaction of the left ventricle to maturation and compaction. The regulated expression of different deiodinases either as part of developmental program or in response to environmental cues allows for active modulation of tissue-specific thyroid hormone signaling. Different studies have shown that models of pathologic ventricular hypertrophy report the re-expression of deiodinase 3(D3) in cardiomyocytes. The stimulation of D3 activity in ventricular hypertrophy and subsequent heart failure is associated with severe impairment of cardiac T3 re-signaling [5].
This case was also associated with adrenal insufficiency, which is most likely due to an isolated glucocorticoid hormone deficiency. The absence of an androgen excess and hypertension excludes deficiency of 11-beta-hydroxlase enzyme while 17-hydrxylase deficiency is excluded by absence of hypertension, hypokalemia plus the fact that it usually presents at puberty. Other possible etiologies include congenital adrenal hypoplasia and familial glucocorticoid deficiency. The findings of high plasma ACTH and high renin with a low cortisol support the diagnosis of congenital adrenal hypoplasia but the absence of the abnormal mutation or deletion in the DAX1 gene in exome sequencing in our baby rules out the common form of disease although autosomal recessive type may occur without gene abnormalities. Familial glucocorticoid deficiency is an isolated defect in glucocorticoid production, which is thought to be related to defective signaling in the ACTH receptor or in the post-ACTH receptor mechanism. It is associated with high levels of ACTH. Alternatively familial glucocorticoid deficiency may be due to an isolated defect in the development of the adrenal zona fasciculata resulting in isolated deficiency of glucocorticoid production. It is unlikely that the noncompaction in this case is related to corticosteroid deficiency because noncompaction of the myocardium is seen in the first four weeks of fetal life whereas the action of the glucocorticoid hormone is exhibited late in pregnancy preparing the fetus for life after birth. Glucocorticoids rise dramatically shortly before birth to help in maturation of fetal tissues and organs [6]. The fetal adrenal gland initiates de novo glucocorticoid synthesis during the second half of gestation at around 28th week of pregnancy. Although glucocorticoid hormone has receptors in the cells (GRs) involved in transcription they do not seem to be contributing to the development of the myocardium in the early days of pregnancy. There is no doubt that institution of hydrocortisone in our case has had a significant effect in reversing the hypotension, which reflects the action of glucocorticoid on the vascular tone.
In conclusion this challenging case shows the importance of of adhering to basic clinical judgment plus correct interpretation of available data and teamwork. Reviewing the literature we think that this is the first report of association of an endocrine defect namely hypothyroidism with noncompaction of the left ventricle.
Disclosure
The authors have nothing to disclose.
Acknowledgement
I am grateful to Professor Guy Fontaine for his valuable comments and his encouragement to publish this work. The authors are thankful to Ms. Bing Borromeo for helping with typing of the manuscript.
References
- Moreira FC, Miglioransa MH, Mautone MP, Müller KR, Lucchese F (2006) Noncompaction of the left ventricle: a new cardiomyopathy is presented to the clinician. Sao Paulo Med J 124: 31-35. [Crossref]
- Almeida AG, Pinto FJ (2013) Non-compaction cardiomyopathy. Heart 99: 1535-1542.
- Ting TW, Jamuar SS, Brett MS, Tan ES, Cham BWM, et al. (2015) Left Ventricular Non-compaction: Is It Genetic. Pediatric Cardiology36:1565-1572.
- Klein I, Danzi S (2007) Thyroid disease and the heart. Circulation 116: 1725-1735. [Crossref]
- Pol CJ, Muller A, Simonides WS(2010) Cardiomyocyte-specific inactivation of thyroid hormone in pathologic ventricular hypertrophy: an adaptative response or part of the problem. Heart Failure Reviews 15: 133-142.
- Rog-Zielinska EA, Richardson RV, Denvir MA, Chapman KE (2014) Glucocorticoids and foetal heart maturation; implications for prematurity and foetal programming. J Mol Endocrinol 52: R125-135. [Crossref]