Abstract
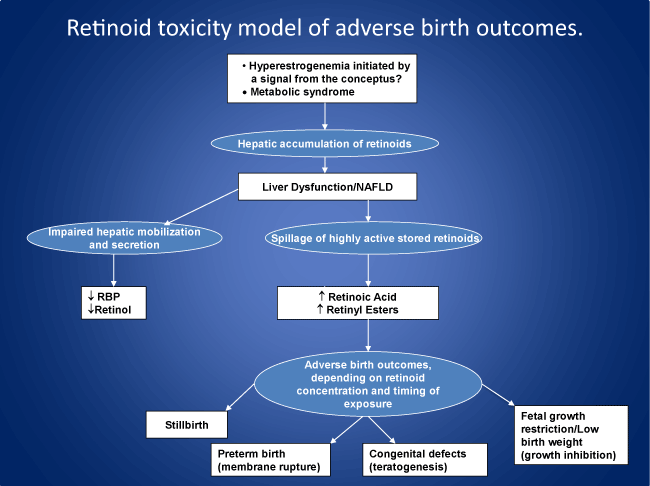
Neither the mechanisms of parturition nor the pathogenesis of preterm birth are well understood. Poor nutritional status has been suspected as a major causal factor, since vitamin A concentrations are low in preterm infants. However, even large enteral doses of vitamin A from birth fail to increase plasma concentrations of vitamin A or improve outcomes in preterm and/or extremely low birthweight infants. These findings suggest an underlying impairment in the secretion of vitamin A from the liver, where about 80% of the vitamin is stored in potentially toxic concentrations. Vitamin A accumulates in the liver and breast during pregnancy in preparation for lactation. While essential in low concentration for multiple biological functions, vitamin A in higher concentration can be pro-oxidant, mutagenic, teratogenic and cytotoxic, acting as a highly surface-active, membrane-seeking and destabilizing compound. Regarding the mechanism of parturition, it is conjectured that by nine months of gestation the hepatic accumulation of vitamin A (retinol) from the liver is such that mobilization and secretion are impaired to the point where stored vitamin A compounds in the form of retinyl esters and retinoic acid begin to spill or leak into the circulation, resulting in amniotic membrane destabilization and the initiation of parturition. If, however, the accumulation and spillage of stored retinoids reaches a critical threshold prior to nine months, e.g., due to cholestatic liver disease, which is common in mothers of preterm infants, the increased retinyl esters and/or retinoic acid rupture the fetal membranes, inducing preterm birth and its complications, including retinopathy, necrotizing enterocolitis and bronchopulmonary dysplasia. Subject to testing, the model suggests that measures taken prior to and during pregnancy to improve liver function could reduce the risk of adverse birth outcomes, including preterm birth.
Key words
liver, retinoids, membranes, parturition, pathogenesis, preterm, birth
Introduction
Every year about 15 million babies are born prematurely and over 1 million children die each year due to complications of preterm birth. Survivors of prematurity often face a lifetime of disability, including learning disabilities and visual and hearing problems; furthermore, rates of preterm birth are rising in almost all countries with reliable data [1]. Defined as a pregnancy ending at less than 37 completed weeks of gestation, preterm birth is the leading cause of infant mortality in the United States and accounts for one-third of deaths in children under one year of age, with an annual societal cost of over $26 billion [2-4].
The occurrence of preterm births rose steadily from 9.4% of all pregnancies in the United States in 1981, to 12.8% in 2006, before declining to 12.7% in 2007 and 12.3% in 2008. Most of the increase was attributed to increases in multiple gestations. In the United States, a pronounced and persistent disparity exists in the rate of preterm birth, with twofold or greater increases among women of African descent compared to other race/ethnic groups. Even after decades of basic science research and public health initiatives, this disparity remains relatively unchanged and unexplained [5].
In high-income countries, 80% of stillbirths are delivered preterm, suggesting that preterm birth and stillbirth share common pathways and mechanisms [6]. Of the 4 million perinatal deaths per year worldwide, up to 99% occur in developing countries [7], with the highest maternal, fetal, and neonatal mortality rates in sub-Saharan Africa and South Asia [8]. Despite massive research effort, the causes and mechanisms of preterm birth and stillbirth remain uncertain and effective interventions are lacking.
Poor nutritional status is suspected of being a major cause of these problems, since vitamin A concentrations are consistently low in preterm infants [9,10]. However, even large enteral doses of vitamin A from birth do not significantly increase plasma concentrations of vitamin A or improve outcomes in preterm or extremely low birthweight infants [11-13]. This failure of supplemention to correct vitamin A concentrations suggests impaired hepatic mobilization and secretion of the carrier protein, retinol-binding protein (RBP); moreover, low retinol (vitamin A alcohol) concentrations do not necessarily indicate a state of vitamin A deficiency and can be associated with hypervitaminosis A [14].
Retinoids
Vitamin A and its congeners (collectively known as retinoids) are mainly dietary-derived fat-soluble signaling molecules that are stored principally in the stellate cells of the liver, from which they are secreted in a regulated process and delivered to the target tissues in the form of retinol-binding protein (RBP). In normal physiological concentrations, retinoids are essential for numerous biological functions such as cellular homeostasis, embryonic development, vision, tissue differentiation, growth, and mucus secretion. In higher concentration, retinoids inhibit cell growth and can be pro-oxidant, cytotoxic, mutagenic, and teratogenic [15-19]. Retinoic acid (RA), the most biologically active metabolite of retinol, is produced from free retinol in a process that involves: 1) hydrolysis of retinyl esters in the liver and the release of retinol into the circulation and its delivery to the target tissues bound to RBP; 2) oxidation of retinol to retinalaldehyde via the action of an alcohol dehydrogenase; and 3) synthesis from retinaldehyde via an aldehyde dehydrogenase reaction, primarily in the cell microsomes. RA exerts its effects by binding to two types of nuclear protein receptors: the retinoic acid receptors (RARs) and retinoid X receptors (rexinoids, RXRs), both of which exist as three distinct gene products (alpha, beta, and gamma). These receptors are members of the steroid/thyroid superfamily of ligand dependent nuclear transcription factors that include the receptors for steroids, thyroid hormone, and vitamin D. Following ligand activation, the receptors function as heterodimeric transcription factors and control the expression of numerous target genes by binding to specific DNA sequences termed RA response elements (RAREs) [20,21].
Given that about 80% of vitamin A is stored in the liver, sudden shifts in these stores to other tissues due to infection, chronic illness or trauma can result in severe vitamin A poisoning. Indeed, an endogenous form of hypervitaminosis A associated with cholestatic liver disease is recognized, due to the spillage of stored retinoids into the circulation in bile [22]. These observations point to the need to reevaluate the role of vitamin A in maternal and child health and in preterm birth in particular.
Retinoid hypothesis of parturition
Neither the mechanisms of preterm birth nor those of normal parturition are well understood. Since a common feature of term and preterm birth is rupture of the fetal membranes, understanding the process of membrane rupture could provide important clues for that of premature rupture of the membranes (PROM) in preterm birth. Preterm PROM (or PPROM) is a leading cause of preterm delivery, accounting for one third of spontaneous preterm births [23] and is strongly associated with adverse pregnancy outcomes [24,25]. Membrane rupture is related to biochemical changes in collagen structure and formation as well as increased oxidative stress [26], involving an imbalance between the synthesis and degradation of collagen within the extracellular matrix of the chorioamniotic membrane induced by matrix metalloproteinases [27]. Pregnancy itself is also associated with oxidative stress [28] and reduced antioxidant capacity [29].
It is proposed that human parturition is due in part to an accumulation of retinoids in the liver in the course of pregnancy, resulting in mild inflammatory changes and the progressive spillage of stored retinoid compounds into the circulation. At around nine months of gestation these compounds have accumulated to the point where they rupture the fetal membranes and initiate parturition. It is hypothesized that early disturbances in liver function and retinoid metabolism, resulting in exposure of the fetus to excess retinoid concentrations, are critically involved in preterm birth and other adverse birth outcomes. Vitamin A can be either antioxidant or pro-oxidant, depending on dosage and cellular condition. At both excessive and therapeutic doses, acute and chronic vitamin A supplementation to laboratory animals leads to increased levels of markers of oxidative stress in liver mitochondria and in the substantia nigra and to alterations in locomotor and exploratory activity [18,19].
Retinoids are essential for the growth and development of the mammalian fetus and placenta [30,31]; for instance, retinoic acid receptor (RAR)-null mutant mice die in utero or shortly after birth and exhibit congenital abnormalities [32]. RARs and retinoid X receptors (RXRs) also show specific spatiotemporal patterns of expression in all developing systems during embryonic development [33]. Indeed, the role of retinoic acid signaling in the genetic regulation of morphogenesis and pattern formation (fetal embryonic development) is well established [34]. The associated amniotic membranes are extra-embryonic structures, indispensable for normal gestation in mammals. The existence of metabolic and molecular pathways of retinoic acid signaling in human fetal membranes is shown by evidence of retinoid receptor (RARα, β, γ and RXRα, β, γ) expression at transcript and protein levels and by enzyme activity involved in the production of retinoic acid [31].
Natural history of pregnancy
Retinol in maternal serum declines during pregnancy, increases at parturition and decreases again with lactation [35]. Retinoids also accumulate in the liver during gestation [36] and in breast tissue [35] in preparation for breastfeeding. It is conjectured that the accumulation of vitamin A in the liver during pregnancy induces a mild form of inflammation and cholestasis, resulting in impaired secretion of RBP and the spillage of active retinoid compounds into the circulation in bile, compounds that are normally excreted via the duodenum. Retinyl esters also leak into the circulation from damaged hepatocytes. The overall result is an endogenous form of vitamin A intoxication associated with an increased percentage of plasma retinyl esters as a fraction of total vitamin A, as well as increased retinoic acid (RA) concentrations. Percent retinyl esters >10% of total vitamin A (retinol plus esters) is an accepted diagnostic criterion of hypervitaminosis A [14]. Liver dysfunction can be precipitated by high concentrations of circulating and stored retinoids and is known to contribute to an endogenous form of vitamin A toxicity through the spillage of retinyl esters and acidic biliary metabolites of vitamin A into the circulation [37,38]. Bile drains from the liver through the gall bladder and common bile duct into the duodenum. In mice, bile contains significantly higher concentrations of retinol (about four times higher) than found in serum and other extrahepatic tissues [39].
It is postulated that retinoids which have accumulated in the liver during the course of pregnancy begin to spill into the circulation as a result of mild hepatic inflammation and cholestasis. As the concentrations of these retinoids steadily rise they begin to weaken and eventually rupture the fetal membranes as a function of their membrane-destabilizing effect, thereby triggering the process of parturition at around nine months of gestation. Although there are no apparent morphologic changes in the liver during normal human pregnancy, functional alterations occur suggesting the presence of inflammatory changes, including a doubling of serum alkaline phosphatase activity as well as increases in serum concentrations of certain bile acids. These observations led to earlier suggestions that pregnancy is associated with sub-clinical cholestasis [40]. In mice, dramatic changes occur in the size of the liver during pregnancy, whereby livers double in weight from the non-pregnant state to day 18 of pregnancy. Growth of the fetal mouse liver begins following implantation and peaks at parturition [41].
With regard to human pregnancy, although little is known about circulating levels of RA in the human fetus and newborn, it is suggested that upon reaching a critical concentration threshold, presumably around nine months of gestation, RA begins to lyse the fetal membranes and induce parturition. Retinol is a highly surface-active, membrane-seeking and destabilizing compound. When unbound to protein, it is known to cause the degradation of the extracellular matrix of chick limb-bone rudiments via effects on lysosomal membranes and the release of lysosomal enzymes [42,43]. Vitamin A can also cause hemolysis of erythrocytes and result in increased permeability and fluidity [44].
The proposed model is supported by a small study of retinol and retinoic acid during pregnancy, showing that retinol concentrations were unrelated to those of the active derivative, all-trans retinoic acid; serum retinol was lower and essentially constant in pregnant women and parturient mothers compared to that in non-pregnant control women. In contrast, all-trans-RA concentrations were higher in the parturient mothers than in control subjects, increasing in the third and fourth month of pregnancy to reach a steady state at about 40% above the initial concentration; moreover, when all-trans-RA concentrations were corrected for changes in binding protein concentrations, i.e., albumin, about a twofold increase occurred in all-trans-RA in the second and third trimesters [45].
Role of retinoids in fetal membranes
The fetal membranes comprise the inner amnion lining of the amniotic cavity and the outer chorion underlying the uterine lining (endometrium). The amnion comprises a single layer of epithelial cells surrounded by connective tissue stroma. These membranes are fundamental for normal gestation in mammals. Amniotic fluid (AF) is produced by the fetus throughout pregnancy, averaging from 500 to 700 ml/day. The fetal membranes are involved in the regulation of AF volume and transfer [46]; this requires aquaporins (AQPs), a family of 13 glycoproteins, which facilitate water flux across cellular membranes. The factors thought to modulate AQP expression in placental and fetal membranes include some members of the nuclear receptor superfamily, e.g., the retinoic acid receptors (RARα, β, γ). RA is reported to regulate APQ1, 3, 5 and 9 in other cellular and tissue environments [47], and the RAR signaling pathway regulates certain target genes in human amniotic membranes [48], but its role in the fetal membranes has remained uncertain. To answer the question of whether RA regulates APQs in the fetal membranes, Sapin and colleagues cultured explants and primary and established amniotic cells to determine which AQPs were transcriptionally modified by all-trans-RA (the most abundant natural form of retinoic acid). Using immunohistochemistry and other methods to determine the impact of all-trans-RA on AQP protein expression and function, they showed that the specific transactivation of a single AQP (that is, AQP3) in the amniotic environment involves RARα. Only RARα (and not RARγ) was able to transduce all-trans-RA signaling on the AQP3 coding gene. These results suggest that upregulation of AQP3 by all-trans-RA participates in maintaining amniotic fluid homeostasis across human fetal membranes. Consistent with the present hypothesis, Sapin and colleagues also concluded that retinoic acid signal dysregulation in fetal membranes could be involved in adverse obstetric outcomes [49].
Role of liver dysfunction and retinoid metabolism in preterm birth
A healthy liver is essential for reproduction in women. Pregnancy in patients with advanced liver disease is uncommon since most women with cirrhosis are infertile and have high rates of anovulation; gestation, if it occurs, carries high risks to both mother and fetus such as increased rates of spontaneous abortion, prematurity, pulmonary hypertension and postpartum hemorrhage. Conversely, after liver transplantation menstruation resumes and most women are able to conceive and give birth [50], although the complication rate is high [51]. Here it is suggested that preterm birth and related adverse birth outcomes occur due to liver dysfunction, when the concentration of retinoids spilled from the liver exceeds a critical threshold.
A central hypothesis of this paper is that liver dysfunction and associated alterations in retinoid metabolism – involving the spillage of stored retinoids into the maternal and fetal circulations in bile, together with the leakage of retinyl esters from damaged hepatocytes – are importantly involved in the pathogenesis of pregnancy complications and adverse birth outcomes. It is proposed that liver dysfunction and endogenous changes in vitamin A metabolism induce a spectrum of disorders, including early pregnancy loss, birth defects, preterm birth, stillbirth, and fetal growth restriction, depending on the concentration of these compounds and the timing of exposure of the fetus to retinoids. Maternal liver dysfunction itself may be due in part to preexisting conditions such as diabetes, obesity or hypertension, or develop in pregnancy associated with gestational diabetes and/or preeclampsia, due to hyperestrogenemia, which can lead to cholestatic liver dysfunction [52].
Membrane rupture and parturition
With regard to the mechanism of preterm birth, it is proposed that cholestatic liver dysfunction and associated increases in circulating concentrations of retinyl esters and/or retinoic acids rupture the fetal membranes, inducing preterm birth and the characteristic features of the preterm infant, including retinopathy, necrotizing enterocolitis, and bronchopulmonary dysplasia. The process leading to preterm birth is conjectured to begin with the accumulation of excess retinoids in the liver in early pregnancy due to hyperestrogenemia, following a signal from the conceptus. Pregnancy increases the sensitivity of the bile ducts to estrogen, and cholestasis often develops during the second and third trimesters of pregnancy [53]. The presence of liver dysfunction is increasingly evident in the progression of preeclampsia to more severe pregnancy-related disorders such as HELLP syndrome (Hemolysis, ELevated liver enzymes, and Low Platelet count). Abnormal liver tests are reported in 10% of pregnancies overall, suggesting reduced liver metabolic capacity in late pregnancy [54], but conventional liver enzyme tests underestimate the true extent of liver dysfunction [55].
The association between liver dysfunction and preterm birth is well documented. For instance, women diagnosed with alcoholic liver disease before birth have an increased risk of preterm birth and of delivering infants small-for-gestational age [56]. Total bile acids are also associated with preterm delivery [57]. After the 12th week of pregnancy, phospholipids, cholesterol and triglycerides increase in response to estrogen stimulation and insulin resistance. Although maternal hypertriglyceridemia appears to have some positive effects, it also increases risk of preeclampsia and preterm birth and may have a role in increasing cardiovascular risk later in life [58]. Consistent with the retinoid hypothesis, hypertriglyceridemia is associated with hypervitaminosis A [59] and is a frequent complication in the therapeutic use
of retinoic acid derivatives [60]. In a study of women with preeclampsia, those who delivered preterm had significantly greater liver enzyme concentrations than those who delivered at term [61]. Menon et al. [62] studied metabolic changes associated with early spontaneous preterm birth (<34 weeks) using high-throughput metabolomics of amniotic fluid (AF) retrieved by transvaginal amniocentesis at the time of labor prior to delivery in an African American population that included a control group of women who delivered at term. They found evidence of altered liver function in many cytochrome P450-related pathways including bile acids, steroids, xanthines, heme, and phase II detoxification of xenobiotics. The largest fold change was in pantothenol, a CoA synthesis inhibitor that was 8-fold more abundant in preterm birth samples. Several bile acids and their metabolites were elevated in preterm birth samples, inversely related to gestational age, including glycocholate, taurocholate, taurochenodeoxycholate, taurodeoxycholate, and glycodeoxycholate. High circulating levels are known to cause fetal stress. Bile acid metabolism, secretion from liver, and liver reuptake can be disrupted by hormonal changes associated with cholestasis in pregnancy [63]. The hypothesis that alterations in liver function and retinoid metabolism are associated with complications of pregnancy and adverse birth outcomes was supported by the results of an animal study carried out by the author and colleagues. The drug streptozotocin was administered to rats early in pregnancy to create an animal model of gestational diabetes and preeclampsia.64 Pup weights of the experimental rats were significantly lower than those of controls (µ=4.6 g vs. 1.4 g, p<0.05), indicating intrauterine growth restriction. Retinoid profiling indicated that retinol concentrations in the experimental dams were significantly lower than in controls. On the other hand, consistent with the model, the percentage of retinyl esters >10% (an indicator of retinoid toxicity) was significantly increased (median, 24% vs. 11%; p=0.008), as was the concentration of retinoic acid (median, 0.0155 vs. 0.0075 µmol/L; p=0.045). Liver enzyme levels were also significantly elevated.
Complications of preterm birth
The proposal that preterm birth is associated with an endogenous form of hypervitaminosis A suggests that the characteristic and usually combined complications of prematurity, e.g., retinopathy, necrotizing enterocolitis and bronchopulmonary dysplasia, are themselves due to hypervitaminosis A. In the case of retinopathy of prematurity, both prenatal and postnatal exposure to isotretinoin (13-cis-retinoic acid, a synthetic vitamin A derivative used in the treatment of severe acne) is known to cause retinopathy and optic nerve abnormalities [65]. Retinoic acid also contributes to light-induced retinopathy in mice via mechanisms that may include plasma membrane permeability and mitochondrial poisoning, leading to caspase activation and mitochondria-associated cell death [66]. Necrotizing enterocolitis (NEC), an inflammatory intestinal disorder primarily seen in premature infants, is characterized by variable damage to the intestinal tract ranging from mucosal injury to full-thickness necrosis and perforation [67]. Although no direct link between retinoid exposure and NEC has been described, adverse side effects attributed to isotretinoin include inflammatory bowel disease as well as depression, suicidality, and teratogenicity [68].
Bronchopulmonary dysplasia (BPD) of the preterm neonate is similar to emphysema of the adult lung in that both are characterized by increased airspace and respiratory insufficiency. They are also strikingly similar in their pathophysiology, including the precipitating effect of oxidative stress, chronic inflammation, increased apoptosis, protease-antiprotease imbalance, elastic fiber deterioration and altered microvascularization [69]. In both diseases there is evidence of low vitamin A concentrations, which have been hypothesized to indicate vitamin A deficiency. However, it has been suggested here that while vitamin A (retinol) concentrations in preterm neonates are low, due to cholestatic liver dysfunction and impaired hepatic mobilization, other vitamin A metabolites are spilled or leaked into the maternal and fetal circulation and induce the characteristic features of the preterm neonate, including bronchopulmonary dysplasia. In support of this hypothesis, maternal vitamin A supplementation during pregnancy and lactation increases oxidative stress parameters in rat neonatal lungs, effects that may be involved in conditions associated with redox dysfunctions and free radical-induced cell damage [70]. Oxidative stress is caused by an imbalance between the production of free radicals and the ability of antioxidant system to detoxify them. These conditions are known as oxygen radical diseases of neonatology and include bronchopulmonary dysplasia, retinopathy, and necrotizing enterocolitis [71].
Thrombocytopenia and anemia are also commonly seen in the preterm infant; for instance, thrombocytopenia occurs in up to a third of preterm neonates admitted to intensive care units [72]. Anemia (hemoglobin <10.5 g/dL) is likewise strongly associated with intrauterine growth restriction and prematurity [73]. Consistent with the model, hypervitaminosis A in infants causes severe thrombocytopenia and anemia, which may be due to a direct inhibitory effect on the growth of all bone marrow cellular components, mediated by the upregulation of cyclin-dependent kinase inhibitors [74].
Conclusion
In 1953 Peter Medawar raised the question: “How does the pregnant mother contrive to nourish within itself, for many weeks or months, a fetus that is an antigenically foreign body?” [75]. In other words, why does the mother fail to reject her fetus as a foreign piece of tissue? The suggestion offered here is that the mother does not reject but rather ejects the fetus, normally at around 37 weeks of gestation, as the result of a biochemical rather than an immunological mechanism. Parturition at term is hypothesized to represent an active process of ejection of the fetus associated with the hepatic accumulation and spillage of stored retinoids due to mild liver dysfunction, which progressively weaken the fetal membranes until they rupture at around the ninth month of gestation. Based on this template for parturition at term, it is further proposed that spontaneous preterm birth is similarly due to more severe liver dysfunction and to concentrations of retinyl esters and retinoic acid that exceed the threshold for inducing rupture of the fetal membranes before 37 weeks of gestation.
The model could be tested by determining whether 1) preterm newborns have significantly lower serum retinol (ROL) concentrations than term newborns, but higher concentrations of retinoic acid (RA), a higher percent retinyl esters (RE), and a higher RE:ROL ratio; 2) mothers of preterm newborns have higher liver enzyme levels than mothers of term neonates; and 3) an increased percentage of REs as a fraction of total serum retinol (>10%) is a strong predictor of preterm birth. Subject to further testing in prospective studies, periodic monitoring of changes in maternal retinoid profiles throughout pregnancy could provide a biomarker for the early identification of impending adverse birth outcomes, which could be useful in prevention.
Approaches to the management of pregnancies resulting in preterm birth and its complications suggested by the retinoid toxicity hypothesis could involve the adoption of measures taken prior to and after conception to improve liver function through dietary and lifestyle changes.
Conflict of interest
The author has a US patent on a “Method for diagnosing gestational diabetes, preeclampsia, and fetal growth restriction.” US Patent Number 8,883,512 B1, November 11, 2014.
http://www.google.com/patents/US8883512
References
- March of Dimes (2013) Low Birthweight. Retrieved March 3, 2016
- Institute of Medicine (2007) Societal Costs of Preterm Birth. The National Academies Press 2007: 396-429. https://www.marchofdimes.com/professionals/25079_1153.asp
- Klebanoff MA, Keim SA (2011) Epidemiology: the changing face of preterm birth. Clin Perinatol 38: 339-350. [Crossref]
- Blencowe H, Cousens S, Chou D, Oestergaard M, Say L, et al. (2013) Born too soon: the global epidemiology of 15 million preterm births. Reprod Health 10 Suppl 1: S2. [Crossref]
- Culhane JF, Goldenberg RL (2011) Racial disparities in preterm birth. Semin Perinatol 35: 234-239. [Crossref]
- Flenady V, Middleton P, Smith GC, Duke W, Erwich JJ, et al. (2011) Stillbirths: the way forward in high-income countries. Lancet 377: 1703-1717. [Crossref]
- Lawn JE, Cousens S, Zupan J; Lancet Neonatal Survival Steering Team (2005) 4 million neonatal deaths: when? Where? Why? Lancet 365: 891-900. [Crossref]
- Bhutta ZA, Yakoob MY, Lawn JE, Rizvi A, Friberg IK, et al. (2011) Stillbirths: what difference can we make and at what cost? Lancet 377: 1523-1538. [Crossref]
- Mactier H, Weaver LT (2005) Vitamin A and preterm infants: what we know, what we don't know, and what we need to know. Arch Dis Child Fetal Neonatal Ed 90: F103-108. [Crossref]
- Abdel Ghany EA, Alsharany W, Ali AA, Younass ER, Hussein JS (2015) Anti-oxidant profiles and markers of oxidative stress in preterm neonates. Paediatr Int Child Health. [Crossref]
- Kennedy KA, Stoll BJ, Ehrenkranz RA, Oh W, Wright LL, et al. (1997) Vitamin A to prevent bronchopulmonary dysplasia in very-low-birth-weight infants: has the dose been too low? The NICHD Neonatal Research Network. Early Hum Dev 49: 19-31. [Crossref]
- Wardle SP, Hughes A, Chen S, Shaw NJ (2001) Randomised controlled trial of oral vitamin A supplementation in preterm infants to prevent chronic lung disease. Arch Dis Child Fetal Neonatal Ed 84: F9-9F13. [Crossref]
- McCauley ME, van den Broek, Dou L, Othman M (2015) Vitamin A supplementation during pregnancy for maternal and newborn outcomes. Cochrane Database Syst Rev 10: CD008666.
- Penniston KL, Tanumihardjo SA (2006) The acute and chronic toxic effects of vitamin A. Am J Clin Nutr 83: 191-201. [Crossref]
- Ganguly J (1989) The Biochemistry of Vitamin A. Boca Raton, FL: CRC Press.
- Hoffman C, Eichele G (1994) Retinoids in development. The Retinoids: Biology, Chemistry, and Medicine. Raven Press, New York Pp: 387–441.
- Litwack G (2007) Vitamin A: Vitamins and Hormones, Elsevier Academic Press, San Diego, USA
- de Oliveira MR, Silvestrin RB, Mello e Souza T, Moreira JCF (2008) Therapeutic vitamin A doses increase the levels of markers of oxidative insult in substantia nigra and decrease locomotory and exploratory activity in rats after acute and chronic supplementation. Neurochem Res 33: 378-383.
- de Oliveira MR (2015) The neurotoxic effects of vitamin A and retinoids. An Acad Bras Cienc 87: 1361-1373. [Crossref]
- Theodosiou M, Laudet V, Schubert M (2010) From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell Mol Life Sci 67: 1423-1445. [Crossref]
- Brun PJ, Yang KJ, Lee SA, Yuen JJ, Blaner WS (2013) Retinoids: Potent regulators of metabolism. Biofactors 39: 151-163. [Crossref]
- Leo MA, Lieber CS (1999) Alcohol, vitamin A, and beta-carotene: adverse interactions, including hepatotoxicity and carcinogenicity. Am J Clin Nutr 69: 1071-1085. [Crossref]
- Goldenberg RL, Culhane JF, Iams JD, Romero R (2008) Epidemiology and causes of preterm birth. Lancet 371: 75-84. [Crossref]
- McParland PC, Taylor DJ (2005) Preterm prelabour rupture of the membranes. Recent Advances in Obstetrics and Gynecology 23: 27-38.
- Menon R (2008) Spontaneous preterm birth, a clinical dilemma: etiologic, pathophysiologic and genetic heterogeneities and racial disparity. Acta Obstet Gynecol Scand 87: 590-600. [Crossref]
- Knudtson EJ, Smith K, Mercer BM, Miodovnik M, Thurnau GR, et al. (2004) Serum homocysteine levels after preterm premature rupture of the membranes. Am J Obstet Gynecol 191: 537-541. [Crossref]
- Fortunato SJ, Menon R (2001) Distinct molecular events suggest different pathways for preterm labor and premature rupture of membranes. Am J Obstet Gynecol 184: 1399-1405. [Crossref]
- Mathews F, Neil A (2005) Antioxidants and preterm prelabour rupture of the membranes. BJOG 112: 588-594. [Crossref]
- Idogun ES, Odiegwu ME, Momoh SM, Okonofua FE (2008) Effects of pregnancy on total antioxidant capacity in Nigerian women. Pak J Med Sci 24: 292-295.
- Wilson JG, Roth CB, Warkany J (1953) An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat 92: 189-217. [Crossref]
2021 Copyright OAT. All rights reserv
- Marceau G, Gallot D, Borel V, Lémery D, Dastugue B, et al. (2006) Molecular and metabolic retinoid pathways in human amniotic membranes. Biochem Biophys Res Commun 346: 1207-1216. [Crossref]
- Lohnes D, Mark M, Mendelshon C, Dollé P, Dierich A (1994) Function of the retinoic acid receptors (RARs) during development. (I) Craniofacial and skeletal abnormalities in RAR double mutants. Development 120: 2723-2748.
- Morriss-Kay GM, Sokolova N (1996) Embryonic development and pattern formation. FASEB J 10: 961-968. [Crossref]
- Maden M (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8: 755-765. [Crossref]
- Bates CJ (1983) Vitamin A in pregnancy and lactation. Proc Nutr Soc 42: 65-79. [Crossref]
- Satre MA, Ugen KE, Kochhar DM (1992) Developmental changes in endogenous retinoids during pregnancy and embryogenesis in the mouse. Biol Reprod 46: 802-810. [Crossref]
- Leo MA, Lieber CS (1985) New pathway for retinol metabolism in liver microsomes. J Biol Chem 260: 5228-5231. [Crossref]
- Shirakami Y, Lee SA, Clugston RD, Blaner WS (2012) Hepatic metabolism of retinoids and disease associations. Biochim Biophys Acta 1821: 124-136. [Crossref]
- Jaensson-Gyllenbäck E, Kotarsky K, Zapata F, Persson EK, Gundersen TE, et al. (2011) Bile retinoids imprint intestinal CD103+ dendritic cells with the ability to generate gut-tropic T cells. Mucosal Immunol 4: 438-447. [Crossref]
- Carter J1 (1991) Serum bile acids in normal pregnancy. Br J Obstet Gynaecol 98: 540-543. [Crossref]
- Dai G, Bustamante JJ, Zou Y, Myronovych A, Bao Q, et al. (2011) Maternal hepatic growth response to pregnancy in the mouse. Exp Biol Med (Maywood) 236: 1322-1332. [Crossref]
- Dingle JT, Fell HB, Goodman DS (1972) The effect of retinol and of retinol-binding protein on embryonic skeletal tissue in organ culture. J Cell Sci 11: 393-402. [Crossref]
- Dingle JT, Lucy JA (1962) Studies on the mode of action of excess of vitamin A. 5. The effect of vitamin A on the stability of the erythrocyte membrane. Biochem J 84: 611-621. [Crossref]
- Urano S, Inomori Y, Sugawara T, Kato Y, Kitahara M, et al. (1992) Vitamin E: inhibition of retinol-induced hemolysis and membrane-stabilizing behavior. J Biol Chem 267: 18365-18370. [Crossref]
- Berggren Söderlund M, Fex GA, Nilsson-Ehle P (2005) Concentrations of retinoids in early pregnancy and in newborns and their mothers. Am J Clin Nutr 81: 633-636. [Crossref]
- Beall MH, van den Wijngaard JP, van Gemert MJ, Ross MG (2007) Regulation of amniotic fluid volume. Placenta 28: 824-832. [Crossref]
- Gao RW, Kong XY, Zhu XX, Zhu GQ, Ma JS, et al. (2015) Retinoic acid promotes primary fetal alveolar epithelial type II cell proliferation and differentiation to alveolar epithelial type I cells. In Vitro Cell Dev Biol Anim 51: 479-487. [Crossref]
- Blanchon L, Marceau G, Borel V, Prat C, Herbet A, et al. (2011) [Implications of retinoid pathway in human fetal membranes: study of target genes]. Gynecol Obstet Fertil 39: 370-372. [Crossref]
- Prat C, Bouvier D, Comptour A, Marceau G, Belville C, et al. (2015) All-trans-retinoic acid regulates aquaporin-3 expression and related cellular membrane permeability in the human amniotic environment. Placenta 36: 881-887. [Crossref]
- Hammoud GM, Almashhrawi AA, Ahmed KT, Rahman R, Ibdah JA (2013) Liver diseases in pregnancy: liver transplantation in pregnancy. World J Gastroenterol 19: 7647-7651. [Crossref]
- Ludvigsson JF, Bergquist A, Ajne G, Kane S, Ekbom A, et al. (2014) A population-based cohort study of pregnancy outcomes among women with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 12: 95-100. [Crossref]
- Forde K, Kaplan DE (2008) Cholestatic liver disease related to systemic disorders. Cholestatic Liver Disease. Humana Press. 135-153.
- Arrese M, Macias RI, Briz O, Perez MJ, Marin JJ (2008) Molecular pathogenesis of intrahepatic cholestasis of pregnancy. Expert Rev Mol Med 10: e9. [Crossref]
- Everson GT (1998) Liver problems in pregnancy: distinguishing normal from abnormal hepatic changes. Medscape Womens Health 3: 3. [Crossref]
- Stefan N, Kantartzis K, Häring HU (2008) Causes and metabolic consequences of Fatty liver. Endocr Rev 29: 939-960. [Crossref]
- Stokkeland K, Ebrahim F, Hultcrantz R, Ekbom A, Stephansson O (2013) Mothers with alcoholic liver disease and the risk for preterm and small-for-gestational-age birth. Alcohol Alcohol 48: 166-171. [Crossref]
- Pata O, Vardarela E, Ozcan A, Serteser M, Unsal I, et al. (2011) Intrahepatic cholestasis of pregnancy: correlation of preterm delivery with bile acids. Turk J Gastroenterol 22: 602-605. [Crossref]
- Ghio A, Bertolotto A, Resi V, Volpe L, Di Cianni G (2011) Triglyceride metabolism in pregnancy. Adv Clin Chem 55: 133-153. [Crossref]
- Ellis JK, Russell RM, Makrauer FL, Schaefer EJ (1986) Increased risk for vitamin A toxicity in severe hypertriglyceridemia. Ann Intern Med 105: 877-879. [Crossref]
- Lilley JS, Linton MF, Fazio S (2013) Oral retinoids and plasma lipids. Dermatol Ther 26: 404-410. [Crossref]
- Phillips JK, Janowiak M, Badger GJ, Bernstein IM (2010) Evidence for distinct preterm and term phenotypes of preeclampsia. J Matern Fetal Neonatal Med 23: 622-626. [Crossref]
- Menon R, Jones J, Gunst PR, Kacerovsky M, Fortunato SJ, et al. (2014) Amniotic fluid metabolomic analysis in spontaneous preterm birth. Reprod Sci 21: 791-803. [Crossref]
- Pathak B, Sheibani L, Lee RH (2010) Cholestasis of pregnancy. Obstet Gynecol Clin North Am 37: 269-282. [Crossref]
- Mawson AR, Bennett W, Tanumihardjo S, Mills J, Smith A, et al. (2010) Altered retinoid profiles in an animal model of preeclampsia, gestational diabetes and fetal growth restriction. Presented at the 57th Annual Meeting of the Society for Gynecological Investigation, Orlando, FL, Reproductive Science 17: 592.
- Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, et al. (1985) Retinoic acid embryopathy. N Engl J Med 313: 837-841. [Crossref]
- Maeda A, Maeda T, Golczak M, Chou S, Desai A, et al. (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem 284: 15173-15183. [Crossref]
- Zani A, Pierro A (2015) Necrotizing enterocolitis: controversies and challenges. F1000Research 4:F1000 Faculty Rev-1373.
- Prevost N, English JC (2013) Isotretinoin: update on controversial issues. J Pediatr Adolesc Gynecol 26: 290-293. [Crossref]
- Bourbon JR, Boucherat O, Boczkowski J, Crestani B, Delacourt C (2009) Bronchopulmonary dysplasia and emphysema: in search of common therapeutic targets. Trends Mol Med 15: 169-179. [Crossref]
- Pasquali MA, Schnorr CE, Feistauer LB, Gelain DP, Moreira JC (2010) Vitamin A supplementation to pregnant and breastfeeding female rats induces oxidative stress in the neonatal lung. Reprod Toxicol 30: 452-456. [Crossref]
- Marseglia L, D'Angelo G, Manti S, Arrigo T, Barberi I, et al. (2014) Oxidative stress-mediated aging during the fetal and perinatal periods. Oxid Med Cell Longev 2014: 358375. [Crossref]
- Chakravorty S, Murray N, Roberts I (2005) Neonatal thrombocytopenia. Early Hum Dev 81: 35-41. [Crossref]
- Breymann C1 (2015) Iron Deficiency Anemia in Pregnancy. Semin Hematol 52: 339-347. [Crossref]
- Perrotta S, Nobili B, Rossi F, Criscuolo M, Iolascon A, et al. (2002) Infant hypervitaminosis A causes severe anemia and thrombocytopenia: evidence of a retinol-dependent bone marrow cell growth inhibition. Blood 99: 2017-2022. [Crossref]
- Medawar PB (1953) Some immunological and endocrinological problems raised by the evolution of viviparity in vertebrates. Sym Soc Exp Biol 7: 320–328.