Abstract
STAT3 is an essential cellular transcription factor that activates a cascade of survival and proliferation signaling program in cells upon cytokine and growth factor stimulus. STAT3 forms a converging point for many upstream activated signaling pathways required for maintaining normal and oncogenic condition. As an active transcription factor, it controls transcription of downstream genes involved in various steps of cancer progression like cell proliferation, migration, immune evasion and angiogenesis. It is known to be constitutively active in many cancers with approximately 40% of breast cancer cases positive for activated STAT3. Apart from the well-studied pY705 activation (canonical pathway), STAT3 is reported to undergo alternative post-translational modifications like pS727 and K685Ac (non-canonical pathway) that are now appearing to be responsible for triggering activated STAT3 in many cancers including breast cancer. Hence, correct designation and targeting ability of these post-translational modifications (PTM) of STAT3 signaling in any particular cancer may hold the key in treating patients with STAT3 overexpression.
Key words
STAT3, non-canonical pathway, STAT3 inhibitors, breast cancer, Her2, SOCS3, PIAS
Introduction
STAT family of proteins
Signal Transducers and Activator of Transcription (STAT) is a family of transcription factors consisting of seven members- Stats 1, 2, 3, 4, 5a, 5b and 6 [1]. The members of STAT family proteins have length of approx. ̴750 to 800 amino acids and a general structure characterized by the presence of various domains like- N-terminal domain, coiled-coil domain, DNA binding domain, SH2 domain and a C-terminal transactivation domain. The N-terminal domain that contain leucine zipper like region is required for interacting with other co-activators like CBP/p300, c-Jun, and Nmi. The coiled-coil domain is required for SH2 domain mediated binding to activated receptor; DNA binding domain is required for recognizing target DNA consensus sequence. The SH2 domain is required for recognizing the pY705 residue on the other STAT molecule for dimer formation and the C-terminal transactivation domain contains conserved Tyr and Ser residues which are keys to activating STAT upon phosphorylation. The members of STAT family are known to either form heterodimer or homodimer to initiate the downstream transcriptional activity upon ligand stimulation [2,3].
Members of STAT family are activated in response to a wide variety of cytokines or growth factors. The general mechanism is that upon binding of the ligand, the receptor (e.g. EGFR, IL-6 receptor) undergoes dimerization followed by auto- or cross-phosphorylation events of tyrosine residues on the receptor. Following receptor activation, STAT protein from the cytoplasm is recruited to the phosphorylated receptor via its SH2 domain leading to phosphorylation of Y705 residue on STAT molecule. Upon phosphorylation, STAT dissociates from receptor and dimerize with another phosphorylated STAT molecule to form an active dimer that can translocate to nucleus and mediates its transcriptional activity [1,4]. STAT proteins are involved in regulation of several normal biological functions like cell differentiation, proliferation, development, apoptosis and inflammation. Genetic knockout studies of individual STAT member have indentified the normal biological function for each of the STAT protein. Loss of STAT1, 2, 4 and 6 majorly led to immune dysfunction [5-7], STAT5a and 5b loss led to developmental defects in mammary glands and lactogenesis [8] while loss of STAT3 alone was found to be embryonically lethal [9]. Among all the members of STAT family, STAT3 and STAT5 are known to overexpress in many type of cancers such as oral, breast, ovarian, head and neck cancer as well as in several hematological malignancies.
Oncogenic role of STAT3 signaling
STAT3, one of the prominent member of STAT family protein is a cytoplasmic factor that relays signal from activated cytokine and growth factor receptor to nucleus where it regulates gene transcription. STAT3 is known to regulate transcription of gene sets involved in cell cycle - e.g. cyclin D1, cyclin E1, and p21; cell survival- e.g. Bcl-2, Bcl-xL and Fas; angiogenesis and metastasis- e.g. VEGF, SNAIL, SLUG etc. [10]. Besides its normal function, STAT3 is also involved in the process of tumorigenesis and transformation. Pioneering work indicating role of STAT3 in oncogenesis by Yu et al. and Cao et al. [11,12] showed that STAT3 is constitutively activated in v-Src transformed cell lines. Subsequently Bromberg et al. showed that constitutive STAT3 activation is required for oncogenic transformation by v-Src and the transfection and expression of constitutively activated form of STAT3 is sufficient to induce transformation of immortalized fibroblast and normal epithelial cell lines derived from either breast or prostate tissue [13]. These findings together indicate that an abnormal STAT3 activity can lead to permanent changes in gene expression programme which in turn can lead to malignant phenotype.
Constitutive activation of STAT3 has been reported in wide range of solid tumors and hematological malignancies including leukemia and lymphoma and it has been used as a prognostic marker to predict disease progression [14]. For instance, in gastric cancer it has been reported that patients with high levels of pY705 STAT3 have a shorter overall survival as compared to patients negative for pY705 STAT3 [15]. Enhanced activation of STAT3 has been reported in case of prostate and ovarian cancer as well [16]. STAT3 expression has been reported to show positive correlation with tumor invasion, lymph node metastasis and tumor grade in colorectal cancer [17]. Increased STAT3 expression has been shown to predict worst clinical outcome for many cancers like cervical cancer [18]. esophageal squamous cell carcinoma [19] and head and neck squamous cell carcinoma [20]. In light of these evidences, it is now clear that the STAT3 is a bonafied mediator of oncogenesis in many human tumors when present in its activated form. Hence blocking or inhibiting STAT3 signaling is considered as a therapeutic target for many cancers.
Regulation of STAT3 signaling
Activation of STAT3 signaling by phosphorylation of pY705 residue is known to be mediated by both receptor tyrosine kinases (EGFR, PDGFR etc.), non-receptor tyrosine kinases (Src, abl kinase) as well as by cytokine receptors such as JAKs [21]. Apart from phosphorylation of Y705 residue, STAT3 also undergoes phosphorylation at S727 residue. The phosphorylation at S727 is known to be mediated by many different kinases such as MAPK, CDK5 etc. [22]. Phosphorylation of STAT3 at both pY705 and pS727 leads to full transactivation of STAT3 signalling [23]. Recently acetylation of STAT3 at K685 residue mediated by CBP/p300 has been reported that also enhances the transcriptional and dimer formation ability of the activated STAT3 molecules [24].
Aberrant and constitutive activation of STAT3 has been observed in many cancers. The major pathway that contributes to abnormally high levels of STAT3 in cancers include, first, excessive STAT3 stimulation achieved by increased secretion of cytokines and growth factors in tumor microenvironment to facilitate paracrine/autocrine signaling of STAT3 in adjacent cells [25] as well as overexpression of protein tyrosine kinases [26,27]. Second, loss of negative feedback loop due to epigenetic alteration and decreased expression level of STAT3 pathway inhibitors such as SOCS3, PTPs and PIAS proteins [28,29]. Third, activating somatic mutations in the SH2 domain of STAT3 such as Y640F, D661H, D661V, D661Y, and N647I increasing the hydrophobicity of SH2 motif to facilitate phosphorylation of Y705 residue and dimerization event [30-32].
Negative regulation of STAT3 signaling
PIAS Protein
PIAS or peptide inhibitor of activated STAT3 is a family of five proteins viz. PIAS1, PIAS3, PIASy, PIASxa, and PIASxb. The major function of PIAS is to inhibit the DNA binding and transcriptional activity of STAT family members. PIAS protein consists of three essential regions- a N terminal LXXLL motif for interacting with nuclear receptors, a serine rich C-terminus and a central ring finger domain required for sumoylation of PIAS binding partners [33]. PIAS proteins are reported to be constitutively expressed within nucleus and continuously repress the function of STAT family proteins. Each of the PIAS family members is known to modulate the function of respective STAT protein for e.g. PIAS1 with STAT1 and p53, PIAS3 with STATs 3, 5a, 5b, and Gfi, PIASx with the androgen receptor and PIASy with LEF1 [34-36]. How PIAS proteins alter the function of transcription factors like STAT3 family, p53 etc. is not clear but recent data suggests that the sumoylation induced by PIAS upon association with its interacting partners could be one of the key modulatory mechanism [33].
PIAS expression was reported to be downregulated while reciprocal increase in STAT3 activation and cell proliferation was observed in glioblastoma [37] Another study in lung squamous cell carcinomas also observed an inverse correlation between PIAS and pSTAT3 expression levels [38]. In case of breast cancer miRNA-21 was reported to inhibit PIAS expression and enhances STAT3 oncogenic signaling [39]. A more thorough study on expression levels and activity of PIAS proteins in breast cancer is required before predicting PIAS as one of the key therapeutic target.
SOCS3 Proteins
Suppressor of cytokine signaling (SOCSs) is family of eight proteins, SOCS 1-7 and CIS. The members of SOCS family consist of an N-terminal domain of variable length, a central SH2 domain required for interacting with pTyr containing domain of JAKs, gp130 and other cytokine receptors and C-terminal SOCS box domain that consists of three alpha helices bound to E3 ubiquitin ligase complex required for binding to signaling proteins and inducing proteasomal degradation [33,32].
The three main mechanisms by which SOCS protein can inhibit STAT3 signaling involves, i) by competing with STAT3 for binding to pTyr motif of activated receptor, ii) by directly binding to JAK receptor and inactivating it, or iii) by binding to signaling protein and targeting it for proteasomal degradation through SOCS box domain [40]. Apart from acting as inhibitor of STAT3 signaling, SOCS3 is also a downstream transcriptional target of STAT3 pathway. Hence upon induction with cytokines or growth factors, the downstream level of SOCS3 increases leading to inhibition of STAT3 by negative feedback loop. However, under normal condition the SOCS3 signaling is under stringent control. In several cancers, the loss of SOCS3 expression due to epigenetic alteration leads to enhanced STAT3 signaling that in turn increases overall cell survival and proliferation [40,41]. In breast cancer patients, loss of SOCS3 expression was considered as a biomarker for poor prognosis and was shown to be associated with increased lymph node metastasis [42]. Recently it has been reported that the promoter of SOCS3 undergoes methylation at CpG island induced by AcK685 STAT3 and DNMT1 interaction in case of TNBC thereby promoting tumor growth [43].
Protein Tyrosine Phosphatases (PTPs)
Phosphorylation is a reversible key event involved in regulating and activating STAT3 signaling in both normal and cancer condition. There are phosphatases present inside the cell that keeps a close check on aberrant and constitutive STAT3 phosphorylation and signaling by deactivation mechanism [21]. The classical PTPs family can be broadly divided into two classes – receptor like PTPs for e.g. CD45 that relies on stimulation by a ligand to initiate dephosphorylation activity and non-transmembrane PTPs including SH2 domain containing SHP1, SHP2, PTP1B (phospho-tyrosine phosphatase 1B), and TC-PTP/ PTPN2 (T cell-protein tyrosine phosphatase) that contain regulatory sequence and catalytic domain controlling their activation. Protein tyrosine phosphatase 1B (PTP1B) is known to dephosphorylate JAK2, TYK2, STAT 5a and STAT 5b, while TC-PTP and SHP2 is known to dephosphorylate JAK2, STAT1, STAT3 and STAT5. PTPMeg2 is a novel phosphatase reported in breast cancer that directly dephosphorylates STAT3. TC45 an isoform of PTPN2 is nuclear localized and regulates the reverse effect of pS727 and pY705 modifications on STAT3 activity. TC45 is activated by the pS727 STAT3 and the activated TC45 in turn dephosphorylate the pY705 residue of STAT3 thereby controlling the pool of specific STAT3 modified form in the cell and the downstream biological function. [44].
Apart from PTPs there is another group of heterogeneous phosphatases called DSPs (dual specific phosphatases). These phosphatases share same mechanism of dephosphorylation as that of classical PTPS, except that their active site is constructed in such a way that it can recognize both phospho-tyr and phospho-Ser/thr residues. Some of the DSPs include MAPK phosphatases (MKPs), KAP (cyclin-dependent kinase associated phosphatase), myotubularins (MTMs) and VH1-related DSP (VHR). Phosphoprotein phosphatase 2A (PP2A) is a Ser/Thr phosphatase whose activity on STAT3 depends upon the STAT3 interacting proteins like SIPAR (STAT3-Interacting Protein as a Repressor) interacts with pY075-STAT3 and promotes dephosphorylation while 14–3-3ζ, protects pS727-STAT3 from PP2A [44,45].
Methods for functional assessment of STAT3 activation
Understanding STAT3 signaling pathway in cancer holds the key to decipher the oncogenic role of STAT3. Development of cellular sensors to monitor STAT3 activity in live cells may allow studies to understand the minor and intricate details of pathway to be captured in normal and disease conditions. Therefore, to have a better understanding of the regulators of STAT3 signaling and to identify potential inhibitors various attempts have been made in developing intracellular sensing technologies that can capture the event of STAT3 signaling in situ using live cells.
The major regulator of STAT3 activation in biological condition is protein tyrosine kinases. There have been efforts in the past where people have developed sensors to monitor activity of protein tyrosine kinases as an assessment to study STAT3 activation. The major breakthrough came with the development of two GFP analogues - CFP (cyan fluorescence protein) and YFP (yellow fluorescence protein) paving the pathway for generation of FRET (fluorescence resonance energy transfer) pair. STAT3 constructs fused to GFP variants- CFP and YFP were prepared and the ability of these fused STAT3 constructs to detect STAT3 dimerization and activity upon cytokine stimulation was monitored using live cell fluorescence spectroscopy and FRET. A two-fold increase in basal FRET signal upon IL-6 treatment was observed as compared to the untreated counterparts indicating increase STAT3 phosphorylation and dimerization [46]. Subsequently in the same year, another STAT3 sensor based on the principle of BRET (Bioluminescence resonance energy transfer) was developed to monitor the preassociation of STAT3 molecules before ligand stimulus in live cells. One STAT3 fused to Rluc (Renilla luciferase)-donor molecule and another STAT3 fused to EYFP (enhanced yellow fluorescent protein)-acceptor molecule was used as BRET partners. It was observed that in the absence of ligand stimulation STAT3 forms less stable complex in the cytoplasm that reorganize to form a more strong dimer upon EGF stimulation as indicated by the increase in BRET ratio [47]. In another study to optically report the phosphorylation of tyrosine residue in STAT3, Trp564 residue of STAT3β was genetically mutated to L-(7-hydroxycoumarin-4-yl) ethylglycine. The 7HC-STAT3β showed increase in fluorescence at 448nm and a second peak at 416 nm upon incubation with Src kinase as compared to only 7HC-STAT3β. To further confirm the phosphorylation of Y705 residue to be responsible for increase in fluorescence intensity, Y705 was mutated to Y705F. The Y705F-7HC-STAT3β had emission peak same as 7HC-STAT3β and the spectrum did not change even upon incubation with Src kinase indicating that the change in fluorescence is an indicator of tyrosine 705 phosphorylation in the SH2 domain of STAT3 [48].
STAT3 oncogene and breast cancer
Role of constitutive STAT3 signaling has been implicated in breast cancer with approx. 50-60% of breast cancer cases found positive for STAT3 overexpression [49]. IL6/gp130/JAK pathway and paracrine signaling is known to be involved in activating and phosphorylating STAT3 in breast cancer as was detected in a panel of breast cancer cell lines [50]. The localization and expression level of phospho STAT3 has been analyzed in many patients based study using different experimental approaches.
The prognostic significance of STAT3 signaling in invasive breast cancer cases was analyzed in a large cohort of patients (n=1270) by monitoring expression level and localization of STAT3 and pY705 STAT3 using IHC in FFPE tissues. Quantification of STAT3 and pY705 STAT3 expression was also done using reverse phase protein array (RRPA) along with analyzing the STAT3 gene expression from the METABRIC cohort. The overexpression of nuclear localized pY705 STAT3 showed a positive correlation with various clinico-pathological parameters like small tumor size, lower grade tumor, negative lymphovascular invasion, hormone receptor positive subtypes and low Ki67 proliferation marker as well as improved breast cancer specific survival (BCSS). While higher STAT3 transcript levels in the METABRIC cohort was observed in the cases with improved BCSS and good prognostic markers, indicating high levels of nuclear pY705 Stat3 as a good prognostic criteria for breast cancer [51]. In another study tissue microarray based analysis of STAT3 and pY705 STAT3 was done in a cohort of 346 node negative breast cancer cases. Out of which 23% of cases had positive STAT3 nuclear expression with significantly improved short term survival while 43.5% cases with positive pY705 STAT3 nuclear expression had significantly improved short term (5 years) and long term (20 years) survival [52]. A recent bioinformatics based analysis of STAT3 gene signature revealed that they are more common in basal-like breast cancers but not in luminal A or luminal B type. The differential gene signatures obtained were more common to immune signaling and inflammation pathway, a phenotype associated with basal like breast cancer but not in luminal A or B [53].
Increasing pool of evidences also highlights the role of STAT3 signaling in maintenance of cancer stem cell population in breast cancer. Breast cancer cell consist of two population type: CD44+CD24– and CD44–CD24+. IHC based analysis for CD44+CD24– and CD44–CD24+ markers was done in a large cohort of invasive and in situ breast carcinomas in which it was found that CD44+ CD24– population is more predominant in basal-like breast cancer while luminal tumors are enriched in CD44–CD24+ type. IL-6/JAK2/STAT3 pathway was found to be active in CD44+CD24– breast cancer cells and treatment with JAK2 inhibitor not only decreased their growth but also significantly blocked the growth of tumor in xenograft model [54,55]. Her2 overexpression increases STAT3 phosphorylation and expression of stem cell markers like Oct-4, Sox-2 and CD44. Knockdown of STAT3 in Her2 overexpressing cells not only decreased the expression of stem cell markers but also diminished the tumor sphere formation ability indicating Her2-STAT3 signaling as a potential mechanism of drug resistance [56,57]. Studies based on tumor samples as well as in cell line models have identified different STAT3 target genes involved in cancer progression. A direct positive correlation between STAT3 expression and levels of survivin [58], Cyclin D1 [59], Twist [60], MMPs [61], HIF1α [62] has been documented in breast cancer patient samples.
Constitutive STAT3 activation and signaling in cancer
pY705 phosphorylation: The Canonical Pathway
STAT3 is known be a latent cytoplasmic transcription factor that gets activated upon binding to its upstream activated kinase receptors followed by phosphorylation of tyrosine 705 residue located in the c-terminal transactivation domain. The pY705 STAT3 then dimerize with another pY705 STAT3 molecule via SH2 doimain-pY705 residues interaction leading to stable dimer formation and translocation to nucleus. The translocation of STAT3 dimers to nucleus is facilitated by importin-α/importin-β1/Ran complex and presence of nuclear localization signal (NLS) [63]. Inside nucleus STAT3 regulate transcription of various downstream targets by binding to the interferon γ DNA consensus sequences (Gamma-Activated Sequence [GAS]) of the promoter region. It regulates transcription of genes involved in various essential cellular functions like cell survival, proliferation, migration, differentiation, etc [64]. The upstream signals that triggers activation of STAT3 signaling via phosphorylation of conserved Y705 residue is known to be as canonical pathway of activation.
Activation of STAT3 via phosphorylation of Y705 residues is known to be mediated by multiple upstream inputs.There are receptors present on plasma membrane like gp130 receptor that lacks intrinsic tyrosine kinase activity and recruits cytoplasmic kinases like JAK family including JAK1, JAK2, JAK3 and TYK2, there are receptors like EGFR, PDGFR, Her2, FGFR, VEGFR, IGFR and HGFR with intrinsic tyrosine kinase activity that itself phosphorylates Y705 residue and induces STAT3 activation [3,31]. Apart from receptors present on plasma membrane there are cytoplasmic kinases also that can activate STAT3 signaling like Bcr-Abl fusion protein, Src kinase family and Bone Marrow X-linked (BMX) kinase [65]. Multiple ligands like cytokines (IL6, LIF, OSM, L-10, and IL-11) and growth factors (EGF, PDGF and CSF-1) are known to stimulate STAT3 signaling. Recently there are many reports that shows that even GPCRs can regulate STAT3 activation and signaling in many cancers by interacting with sphingosine-1-phosphate receptor-1 (SPR-1) (Figure 1) [66].
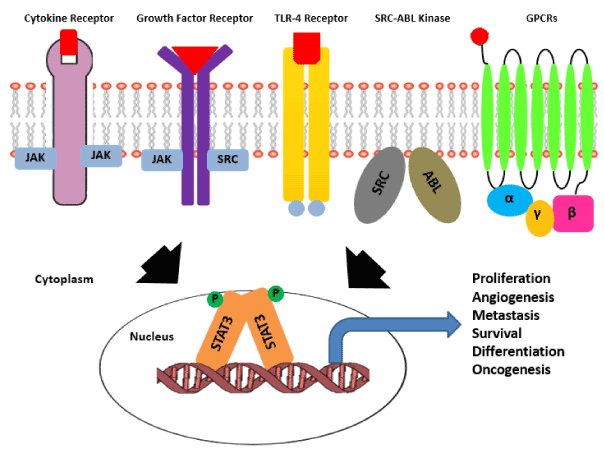
Figure 1: Pathways activating STAT3 signaling in cancer. Activation of STAT3 signaling is achieved by diverse set of upstream receptors known to be involved in cancer progression.There are receptor tyrosine kinases with intrinsic tyrosine kinase activity like gp130 receptor and TLR (Toll like receptors), there are growth factor receptors that lack intrinsic kinase activity and depend on the cytoplasmic kinases for the same. There is a class of soluble ser/thr kinases like Src-Abl that can also activate cytoplasmic STAT3. Unlike RTKs and non-RTKs, GPCRs get activated by guanine nucleotide exchange and in turn activate downstream STAT3 signaling cascade. In all the conditions the cytoplasmic STAT3 molecule gets activated via phosphorylation, followed by homodimerization and nuclear translocation of the STAT3 dimer. Inside nucleus STAT3 dimer switches -on the transcriptional program essential for various aspects of cancer progression.
The canonical pathway of STAT3 signaling is known to be responsible for regulating expression of genes involved in various aspects of cancer progression like immune evasion, angiogenesis, proliferation, inhibition of apoptosis etc. For many years’ activation of STAT3 pathway in cancers is detected by the presence of pY705 form of STAT3. pY705 STAT3 has been used as a biomarker marker for breast cancer, colon cancer, osteosarcoma and many other cancers for predicting the disease prognosis and overall survival. Hence inhibitors have been developed that either targets the upstream receptor or directly interferes with the STAT3 dimers to inhibit canonical STAT3 signaling. Earlier it was presumed that the normal and oncogenic functioning of STAT3 majorly relies on its canonical pathway of activation. But recently there are many reports that have come up highlighting existence of an alternative pathway of STAT3 activation that may or may not depend upon the classical canonical STAT3 pathway [67].
pS727 Phosphorylation and K685 Acetylation of STAT3: The Non Canonical Pathway
The non-canonical pathway of STAT3 signaling has been implicated to STAT3 function independent of classical Y075 phosphorylation. Data from recent literature shows that, STAT3 undergoes phosphorylation at another crucial residue i.e. S727. All the members of STAT family except for STAT2 are known to undergo phosphorylation at S727 residue. The S727 residue is located at the C-terminal transactivation domain region of STAT3 approx. 22 amino acids away from Y705 residue [68]. There are many converging kinases known to be responsible for phosphorylating STAT3 at S727 like MAPK, JNK, PKC family including PKC-δ, PKC-ε as well as CDK5 [22,69]. With the initial discovery, it was thought the phosphorylation of STAT3 at S727 along with pY705 is required for full activation of STAT3[70]. Subsequently many reports highlighted the independent role of pS727 and pY705 in regulating STAT3 function in cancer. It either has a reciprocal inhibitory effect or may have an opposite effect on pY705 STAT3 function.
In case of melanoma [71] and chronic lymphocytic leukemia [72] constant activation of STAT3 pathway was observed with pS727 being detected in almost all the cases while very few of the samples had pY705 STAT3 present. This phosphorylation of STAT3 at S727 residue was important for the survival as well as the transcriptional activity of the cells. In case of glioblastoma (GBM) high levels of pS727 STAT3 leads to shorter progress free survival [73]. Increased level of pS727 STAT3 in GMB cell lines correlates with the increased intrinsic radioresistance nature and treatment with Gö6976 inhibitor induces radiosensitization in those GBM cell lines that had high levels of pS727 STAT3 and no or weak expression of pY705 STAT3 [74]. The differential role of pS727 and pY705 STAT3 is also seen in deciding the fate of mouse embryonic stem cells (mESC). While pY705 STAT3 is absolutely essential for mESC self-renewal, pS727 majorly regulates the transition of mESC from pluripotent state to neuronal differentiation [75]. Upon DNA damage with Topoisomerase I inhibitor in case of colorectal cancer, cdk5 associates with and phosphorylates STAT3 at S727 in the absence of pY705 and induces expression of DNA repair gene Eme1, suggesting an alternative intrinsic resistance mechanism to chemotherapy [76,77].
A significant correlation was also seen between the pS727 STAT3 expression level and ER negative status of breast cancer in both cell lines and patient samples. pS727 STAT3 showed positive correlation with both tumor size and cancer stage while no such correlation was observed for pY705 STAT3. Knockdown of ER expression using siRNA increases pS727 expression in ER positive breast cancer cell lines indicating that ER is either directly or indirectly regulating STAT3 expression [78]. Recently the significance of STAT3 function located in mitochondria has also been highlighted. The mitochondrial localized STAT3 increases the activity of ETC components like succinate oxidoreductase (complex II), ATP synthase (complex V) and lactate dehydrogenase thereby regulating the cellular respiration in cancer. In case of breast cancer the predominant pS727 STAT3 localized in mitochondria promotes tumor growth and metastasis by inhibiting production of reactive oxygen species as compared to the pY705 STAT3 [79].
Apart from phosphorylation at S727 position, STAT3 also undergoes acetylation of lysine residue located at 685 amino acid position in the SH2 domain of STAT3. The acetylation of STAT3 is induced by interaction of p300/CBP histone acetyl transferase protein with the C-terminal domain of STAT3 that further increases upon stimulus with IL6 or IL6 and p300/CBP both [23]. Upon acetylation of K685 residue STAT3 shows increased nuclear localization, DNA binding ability and enhanced transactivation activity. Yuan et al. (2005) for the first time reported that acetylation of lysine685 residue is crucial for STAT3 to form stable dimers and regulate gene transcription that took place even in the absence of pY075 and pS727 residues [24]. The acetylation effect of p300 is reduced upon treatment with HDAC 1 or 2. While loss of K685 acetylation resulted in decreased expression of essential STAT3 downstream genes regulating cell proliferation and survival like cyclin D1, c-myc and Bcl-xL.
Enhanced expression of acetylated form of STAT3 has been reported in case of melanoma, colorectal cancer, ovarian cancer [80], lung cancer as well as in case of triple negative breast cancer as compared to the adjacent normal tissue sample detected using immunohistochemistry [43]. The increased level of acetylated form of STAT3 promotes tumor growth in vivo while K685R mutant expressing cells showed significantly slower tumor growth. Acetylated K685 STAT3 was reported to interact with DNMT1 and methylates the CpG islands in the promoter region of tumor suppressor genes like CDKN2A, DLEC1, STAT1, TP53, SHP-1 and SOCS3 thereby decreasing their expression and tumor suppressive function. K685 acetylated STAT3-DNMT1 interaction also inhibits the transcription of ERα by methylating the promoter in case of TNBC cell lines. Treatment with resveratrol (a histone deacetylase activator) significantly decreases the STAT3 K685 acetylation followed by increase in ERα expression at both protein and mRNA level as well as sensitizes the TNBC cells to tamoxifen-induced cell death [43]. Acetylated K685 STAT3 is also known to interact with CD44 and regulate transcription of cyclin D1 gene in case of gastric cancer [81].
Acetylated STAT3 is also known to regulate the non-canonical NF-κB signaling pathway. Activation of STAT3 in presence of p300/CBP enhances STAT3 K685 acetylation thereby activating STAT3. The acetylated STAT3 in turn activates IKKα kinase that leads to phosphorylation of two key serine residues in C- terminal region of p100 required for processing of p100 to p52. Overexpression of p52 protects cells from undergoing apoptotic pathway. Overexpressing the K685R acetylation mutant significantly reduced the processing of p100 to p52 thereby inducing cell death. Hence the interaction of acetylated K685 STAT3 and processing of p100 to p52 provides an alternative survival pathway that cancer cells might adopt to escape therapy [82,83]. There are also reports that highlight that uSTAT3 (unphosphorylated STAT3) interacts with NFκB and forms a complex that is translocated to nucleus via interaction with importin-α3. Inside nucleus the uSTAT3 and NFκB complex regulate transcription of genes such as RANTES, IL6, IL8, MET, and MRAS (Figure 2) [84].
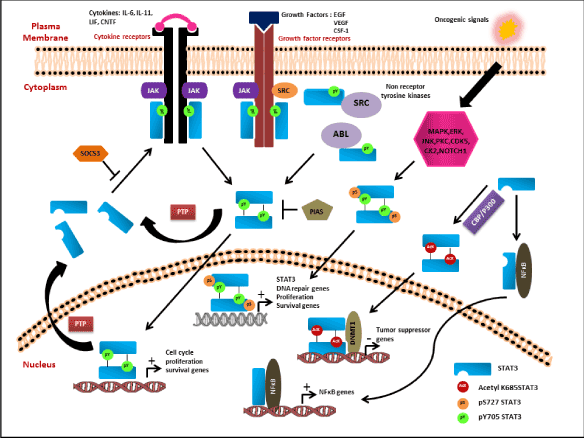
Figure 2: Classical Canonical and Non-Canonical Pathway of STAT3 Activation. The canonical pathway of STAT3 is activated by both receptor tyrosine kinases and non-receptor tyrosine kinases upon stimulus with various growth factors or cytokines. The activated receptors phosphorylate the Y705 residue of Stat3 leading to homodimerization and nuclear translocation of the dimers followed by increase in transcription of cell cycle, proliferation, cell survival genes. The active Stat3 signaling is constantly kept under check via various negative
feedback loops like SOCS3, PIAS and PTPs to inhibit the active pathway. The S727 residue of Stat3 is known to be activated in response to various upstream kinases (MAPK, ERK, CDK5, CK2) and some unknown mechanism. pS727 either alone or together with pY705 forms the non-canonical pathway that downstream activates genes involved in cell proliferation, survival, DNA repair, drug resistance and some unknown genes. Apart from phosphorylation of S727 residue, STAT3 also undergoes acetylation at K685 residue that induces Stat3 homodimerization even in the absence of pY075 and pS727. The acetylated Stat3 dimers interact with DNMT1 and methylate the promoter of tumor suppressor genes like CDKN2A, DLEC1, STAT1, TP53,
SHP-1 and SOCS3 to inhibit its expression. The non-phospho form of STAT3 is known to interact and form complex with NFκB to downstream activate the transcription of NFκB related genes.
STAT3 is also known to undergo reversible methylation at K140 residue. Set9 a histone methyl transferase methylates STAT3 while LSD1 (lysine specific demethylase 1) demethylates it. Methylation at K140 residue is a nuclear event that is secondary to S727 phosphorylation. This particular modification is known to decrease the DNA binding and transcriptional activity of STAT3 molecule [85].
STAT3 Inhibitors
It is a well-known fact that STAT3 is widely present in tumors and its effective functional inhibition could prove to be a valuable anti-cancer strategy. This has led to lot of research on identifying STAT3 targeting compounds. Several STAT3 targeting strategies have been reported. Inhibitors that target at multiple steps of STAT3 activation have been developed. Various cell surface receptor inhibitors such as the peptide aptamer KDI1 and small molecule PD153035 interact with EGFR and inhibit phosphorylation of STAT3 at Y705 preventing its activation and dimerization. FGFR inhibition with Ponatinib decreases both STAT3 phosphorylation and tumor growth in vivo [86]. Short peptides derived from the helices of N-terminal domain of STAT3 protein binds to it and leads to inhibition of transcriptional activity. In another approach, in order to block STAT3 activity, STAT3 dimerization is targeted by the drug development research community. STAT3 dimerization blockers that target blocking the SH2 domain of STAT3 are Stattic, S3I-M2001 and STAT3 inhibitory peptides such as 15-DPP, rPP-C8, LLL12, FLLL31, and FLLL32. These compounds inhibit the formation of active dimers and therefore inhibit STAT3 activation [87]. Inhibitors that dephosphorylate STAT3 like resveratrol, cisplatin, curcumin are also present. G quartret oligodeoxynucleotides inhibit STAT3 at micromolar concentrations but its use is problematic due to the large size and potassium dependence of the compound, which limits cellular delivery. Decoy oligodeoxynucleotides (dODNs) have short stretches of dsDNA, containing transcription factor consensus binding sequence [31]. Cells which show constitutive expression of STAT3, treatment with STAT3 dODNs induce cell death by preventing its nuclear translocation in such cases.
A school of thoughts has stated STAT3 as an ‘undruggable target’ and thus aimed at targeting JAK kinases to inhibit activated STAT3 signaling. Various inhibitors that target JAK kinases like Ruxolitinib, Tofacitinib, Momelotinib, Fedratinib etc. [3] have been evaluated. JAK inhibitors such as WP-1066, LS-104, and CEP-701 continue into clinical trials but some of them were discontinued due to their adverse effects [40]. Inhibiting STAT3 activation by targeting JAKs is also questionable as JAK inhibition also alters other downstream protein targets. Therefore, development of inhibitors that directly target STAT3 activation remained as a complicated and challenging task. To summarize, STAT3 is an attractive target for cancer therapy with clear evidences on its role in cancer progression. Drugs that target STAT3 are yet to be accomplished in future which aim at various STAT3 upstream and downstream activators. Some of the inhibitors that target STAT3 are in clinical trials as indicated in (Table 1). However, it is challenging to identify direct inhibitors of STAT3, but further in depth analysis of molecular variants and their role in regulation of STAT3 downstream signaling will pave the way of developing novel inhibitors.
Inhibitor |
IC50 Value
|
Mode of inhibition |
Clinical trial phase |
Reference |
Ruxolitinib |
JAK1=2.7nM
JA JAK2=4.5nM |
JAK1/2 Inhibitor |
Phase II |
[88] |
Tofacitinib |
1nM |
JAK Inhibitor |
Phase III |
[89] |
Baricitinib |
JAK1=5.9 nM
JAK2=5.7 nM |
JAK1/2 Inhibitor |
Phase III |
[90] |
Momelotinib |
JAK1=11nM
JAK2=18 nM |
JAK1/2 Inhibitor |
Phase III |
[91] |
Filgotinib |
JAK1=10 nM
JAK2=28 nM,
JAK3=810nM TYK2=116nM |
JAK1 Inhibitor |
Phase II |
[92] |
Pacritinib |
23nM |
JAK Inhibitor |
Phase II |
[93] |
Auranofin |
3.37 μM |
STAT3 Phosphorylation |
Phase II |
[94] |
Indirubin |
5 μM |
Inhibits phosphorylation of STAT3 |
- |
[95] |
|
|
3 μM |
Suppresses STAT3 activation |
_ |
[96] |
Stattic |
5.1 μM |
Suppresses STAT3 activation and nuclear translocation |
_ |
[97] |
Niclosamide |
0.7 μM |
Activation, nuclear translocation and transactivation of STAT3 |
PHASE II |
[98] |
Cerdulatinib
|
JAK1=12nM
JAK2=6 Nm
JAK3=8 nM |
JAK Inhibitor
|
PHASE II |
[99] |
FLLL32 |
<5 μM |
JAK2/STAT3 inhibitor |
- |
[100] |
S31-201 |
86 μM
|
Inhibits STAT3 Dimerization |
- |
[101] |
Curcumin |
15.9μM |
Inhibits Phosphorylation Of STAT3 |
PHASE I/II/III |
[102] |
3,3′-diindolyl-methane |
17 μM |
Inhibits phosphorylation of STAT3 |
PHASE I/II/III |
[103] |
Oleanolic acid/CDDO - Me |
4 μM |
Inhibits Phosphorylation Of STAT3 |
PHASE I/II/III |
[104] |
AZD1480 |
0.26 nM |
JAK2 Inhibitor |
PHASE I |
[105] |
Table 1. List of potential STAT3 inhibitors currently employed in clinical trial for various cancers
Conclusion
Increasing pieces of research evidences suggests that apart from classical canonical pathway of activation (pY705 STAT3) that is very well established in breast and other cancers, non-canonical signaling (pS727 and K685Ac) plays equivocal role in activating and regulating STAT3 functions. The significance of pS727 STAT3 in CLL, melanoma, breast cancer as well as in lung cancer shows that the STAT3 pathway can function independently of pY705 phosphorylation in controlling cell survival and tumor growth. K685 acetylation of STAT3 can independently induce STAT3 dimerization even in the absence of pY705 residue. The stable dimers formed can interact with DNMT1 to suppress expression of tumor suppressor genes and favour growth of tumor cells. It is quite possible that different post-translational modifications of STAT3 might be altering the transcriptional preference of STAT3 dimers and regulates a different set of transcriptional programme that can be stimulus dependent. Hence, in the light of the above evidences it is now clear that STAT3 pY705 should no longer be considered as sole signature of STAT3 activation but require to consider and evaluate other modifications as equally important PTM markers for STAT3 target. This will not only help in targeting the right population of activated STAT3, but it will also help in correlating different PTM forms of this target with specific disease condition and might prove to be a promising drug target in future.
Acknowledgement
We gratefully acknowledge institutional facility support for this work. Research support from DBT, New Delhi, India (BT/PR3651/MED/32/210/2011) is acknowledged.
References
- Darnell JE, Kerr IM, Stark GR (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415-1421.
- Darnell JE (1997) STATs and gene regulation. Science 277: 1630-1635. [Crossref]
- Xiong A, Yang Z, Shen Y, Zhou J, Shen Q (2014) Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 6: 926-957.
- Ihle JN (2001) The Stat family in cytokine signaling. Current opinion in cell biology 13: 211-217.
- Durbin JE, Hackenmiller R, Simon MC, Levy DE (1996) Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84: 443-450.
- Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. (1996) Essential role of Stat6 in IL-4 signaling. Nature 380: 627-630.
- Kaplan MH, Sun YL, Hoey T, Grusby MJ (1996) Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382: 174-177.
- Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L (1997) Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes & development 11: 179-186.
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N et al. (1997) Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proceedings of the National Academy of Sciences of the United States of America 94: 3801-3804.
- Huang S (2007) Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clinical cancer research: an official journal of the American Association for Cancer Research 13: 1362-1366.
- Cao X, Tay A, Guy GR, Tan YH (1996) Activation and association of Stat3 with Src in v-Src-transformed cell lines. Molecular and cellular biology 16: 1595-1603.
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, et al. (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269: 81-83.
- Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE (1998) Stat3 activation is required for cellular transformation by v-src. Molecular and cellular biology 18: 2553-2558.
- Bournazou E, Bromberg J (2013) Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT 2: e23828. [Crossref]
- Xiong H, Du W, Wang JL, Wang YC, Tang JT, Hong J, et al. (2012) Constitutive activation of STAT3 is predictive of poor prognosis in human gastric cancer. Journal of molecular medicine 90: 1037-1046.
- Garcia R, Yu CL, Hudnall A, Catlett R, Nelson KL, Smithgall T, et al. (1997) Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 8: 1267-1276.
- Kusaba T, Nakayama T, Yamazumi K, Yakata Y, Yoshizaki A, Inoue K, et al. (2006) Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncology reports 15: 1445-1451.
- Takemoto S, Ushijima K, Kawano K, Yamaguchi T, Terada A, Fujiyoshi N, et al. (2009) Expression of activated signal transducer and activator of transcription-3 predicts poor prognosis in cervical squamous-cell carcinoma. British journal of cancer 101: 967-972.
- Schoppmann SF, Jesch B, Friedrich J, Jomrich G, Maroske F, Birner P (2012) Phosphorylation of signal transducer and activator of transcription 3 (STAT3) correlates with Her-2 status, carbonic anhydrase 9 expression and prognosis in esophageal cancer. Clinical & experimental metastasis 29: 615-624.
- Macha MA, Matta A, Kaur J, Chauhan SS, Thakar A, Shukla NK, et al. (2011) Prognostic significance of nuclear pSTAT3 in oral cancer. Head & neck 33: 482-489.
- Banerjee K, Resat H (2016) Constitutive activation of STAT3 in breast cancer cells: A review. International journal of cancer 138: 2570-2578.
- Decker T, Kovarik P (2000) Serine phosphorylation of STATs. Oncogene 19: 2628-2637.
- Wang R, Cherukuri P, Luo J (2005) Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. The Journal of biological chemistry 280: 11528-11534.
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307: 269-273.
- Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9: 798-809. [Crossref]
- Quesnelle KM, Boehm AL, Grandis JR (2007) STAT-mediated EGFR signaling in cancer. Journal of cellular biochemistry 102: 311-319.
- Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA (2006) Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer research 66: 3162-3168.
- Shuai K (2006) Regulation of cytokine signaling pathways by PIAS proteins. Cell Res 16: 196-202. [Crossref]
- Croker BA, Kiu H, Nicholson SE (2008) SOCS regulation of the JAK/STAT signaling pathway. Seminars in cell & developmental biology 19: 414-422.
- Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. (2012) Somatic STAT3 mutations in large granular lymphocytic leukemia. The New England journal of medicine 366: 1905-1913.
- Johnston PA, Grandis JR (2011) STAT3 signaling: anticancer strategies and challenges. Mol Interv 11: 18-26. [Crossref]
- Zhang HF, Lai R (2014) STAT3 in Cancer-Friend or Foe? Cancers (Basel) 6: 1408-1440. [Crossref]
- Clevenger CV (2004) Roles and regulation of stat family transcription factors in human breast cancer. The American journal of pathology 165: 1449-1460.
- Liu B, Gross M, ten2021 Copyright OAT. All rights reservional corepressor of Stat1 with an essential LXXLL signature motif. Proceedings of the National Academy of Sciences of the United States of America 98: 3203-3207.
- Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. (1997) Specific inhibition of Stat3 signal transduction by PIAS3. Science 278: 1803-1805.
- Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, et al. (1998) Inhibition of Stat1-mediated gene activation by PIAS1. Proceedings of the National Academy of Sciences of the United States of America 95: 10626-10631.
- Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, et al. (2008) Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clinical cancer research: an official journal of the American Association for Cancer Research 14: 4694-4704.
- Kluge A, Dabir S, Vlassenbroeck I, Eisenberg R, Dowlati A (2011) Protein inhibitor of activated STAT3 expression in lung cancer. Molecular oncology 5: 256-264.
- Wang Z, Han J, Cui Y, Zhou X, Fan K (2013) miRNA-21 inhibition enhances RANTES and IP-10 release in MCF-7 via PIAS3 and STAT3 signaling and causes increased lymphocyte migration. Biochemical and biophysical research communications 439: 384-389.
- Kamran MZ, Patil P, Gude RP (2013) Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int 2013: 421821. [Crossref]
- Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, et al. (2005) SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene 24: 6699-6708.
- Nakagawa T, Iida S, Osanai T, Uetake H, Aruga T, Toriya Y, et al. (2008) Decreased expression of SOCS-3 mRNA in breast cancer with lymph node metastasis. Oncology reports 19: 33-39.
- Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. (2012) Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proceedings of the National Academy of Sciences of the United States of America 109: 7765-7769.
- Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nature reviews. Molecular cell biology 7: 833-846.
- Ren F, Geng Y, Minami T, Qiu Y, Feng Y, Liu C, et al. (2015) Nuclear termination of STAT3 signaling through SIPAR (STAT3-Interacting Protein as a Repressor)-dependent recruitment of T cell tyrosine phosphatase TC-PTP. FEBS letters 589: 1890-1896.
- Kretzschmar AK, Dinger MC, Henze C, Brocke-HK, Horn F (2004) Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. The Biochemical journal 377: 289-297.
- Schroder M, Kroeger KM, Volk HD, Eidne KA, Grutz G (2004) Preassociation of nonactivated STAT3 molecules demonstrated in living cells using bioluminescence resonance energy transfer: a new model of STAT activation? Journal of leukocyte biology 75: 792-797.
- Lacey VK, Parrish AR, Han S, Shen Z, Briggs SP, Ma Y, et al. (2011) A fluorescent reporter of the phosphorylation status of the substrate protein STAT3. Angewandte Chemie 50: 8692-8696.
- Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, et al. (2007) Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res 9: R32. [Crossref]
- Lieblein JC, Ball S, Hutzen B, Sasser AK, Lin HJ, et al. (2008) STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 8: 302. [Crossref]
- Aleskandarany MA, Agarwal D, Negm OH, Ball G, et al. (2016) The prognostic significance of STAT3 in invasive breast cancer: analysis of protein and mRNA expressions in large cohorts. Breast Cancer Res Treat 156: 9-20. [Crossref]
- Dolled FM, Camp RL, Kowalski DP, Smith BL, Rimm DL (2003) Tissue microarray analysis of signal transducers and activators of transcription 3 (Stat3) and phospho-Stat3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clinical cancer research: an official journal of the American Association for Cancer Research 9: 594-600.
- Tell RW, Horvath CM (2014) Bioinformatic analysis reveals a pattern of STAT3-associated gene expression specific to basal-like breast cancers in human tumors. Proceedings of the National Academy of Sciences of the United States of America 111: 12787-12792.
- Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. (2011) The JAK2/STAT3 signaling pathway is required for growth of CD44(+) CD24 (-) stem cell-like breast cancer cells in human tumors. The Journal of clinical investigation 121: 2723-2735.
- Wang X, Wang G, Zhao Y, Liu X, Ding Q, Shi J, et al. (2012) STAT3 mediates resistance of CD44(+) CD24(-/low) breast cancer stem cells to tamoxifen in vitro. Journal of biomedical research 26: 325-335.
- Kim JS, Kim HA, Seong MK, Seol H, Oh JS, Kim EK, et al. (2016) STAT3-survivin signaling mediates a poor response to radiotherapy in HER2-positive breast cancers. Oncotarget 7: 7055-7065.
- Chung SS, Giehl N, Wu Y, Vadgama JV (2014) STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. International journal of oncology 44: 403-411.
- Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, et al. (2006) Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clinical cancer research: an official journal of the American Association for Cancer Research 12: 11-19.
- Liu LC, Su CH, Wang HC, Chang WS, Tsai CW, et al. (2014) Contribution of personalized Cyclin D1 genotype to triple negative breast cancer risk. Biomedicine (Taipei) 4: 3. [Crossref]
- Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, et al. (2007) Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer research 67: 9066-9076.
- Song Y, Qian L, Song S, Chen L, Zhang Y, Yuan G, et al. (2008) Fra-1 and Stat3 synergistically regulate activation of human MMP-9 gene. Molecular immunology 45: 137-143.
- Pawlus MR, Wang L, Hu CJ (2014) STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 33: 1670-1679.
- Cimica V, Chen HC, Iyer JK, Reich NC (2011) Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-β1. PLoS One 6: e20188. [Crossref]
- Levy DE, Lee CK (2002) What does Stat3 do? The Journal of clinical investigation 109: 1143-1148.
- Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, et al. (2011) Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer cell 19: 498-511.
- Ram PT, Iyengar R (2001) G protein coupled receptor signaling through the Src and Stat3 pathway: role in proliferation and transformation. Oncogene 20: 1601-1606.
- Sellier H, Rébillard A, Guette C, Barré B, Coqueret O (2013) How should we define STAT3 as an oncogene and as a potential target for therapy? JAKSTAT 2: e24716. [Crossref]
- Wen Z and Darnell JE (1997) Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic acids research 25: 2062-2067.
- Tkach M, Rosemblit C, Rivas MA, Proietti CJ, Flaque MCD, Mercogliano MF, et al. (2013) p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocrine-related cancer 20: 197-212.
- Wen Z, Zhong Z, Darnell JE (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82: 241-250.
- Sakaguchi M, Oka M, Iwasaki T, Fukami Y, Nishigori C (2012) Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. The Journal of investigative dermatology 132: 1877-1885.
- Hazan HI, Harris D, Liu Z, Liu J, Li P, Chen X, et al. (2010) STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 115: 2852-2863.
- Lin GS, Chen YP, Lin ZX, Wang XF, Zheng ZQ, Chen L (2014) STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. International journal of clinical and experimental pathology 7: 3141-3149.
- Ouédraogo ZG, Müller BM, et al. (2016) STAT3 Serine 727 Phosphorylation: A Relevant Target to Radiosensitize Human Glioblastoma. Brain Pathol 26: 18-30. [Crossref]
- Huang G, Yan H, Ye S, Tong C, Ying QL (2014) STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem cells 32: 1149-1160.
- Courapied S, Sellier H, de Carne TS, Vigneron A, Bernard AC, Gamelin E, et al. (2010) The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. The Journal of biological chemistry 285: 26765-26778.
- Vigneron A, Gamelin E, Coqueret O (2008) The EGFR-STAT3 oncogenic pathway up-regulates the Eme1 endonuclease to reduce DNA damage after topoisomerase I inhibition. Cancer research 68: 815-825.
- Yeh YT, Ou-Yang F, Chen IF, Yang SF, Wang YY, Chuang HY, et al. (2006) STAT3 ser727 phosphorylation and its association with negative estrogen receptor status in breast infiltrating ductal carcinoma. International journal of cancer. Journal international du cancer 118: 2943-2947.
- Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, et al. (2013) Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. The Journal of biological chemistry 288: 31280-31288.
- Li J, Cui G, Sun L, Wang SJ, Li YL, Meng YG, et al. (2013) STAT3 acetylation-induced promoter methylation is associated with downregulation of the ARHI tumor-suppressor gene in ovarian cancer. Oncology reports 30: 165-170.
- Lee JL, Wang MJ, Chen JY (2009) Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. The Journal of cell biology 185: 949-957.
- Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. (2009) Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer cell 15: 283-293.
- Nadiminty N, Lou W, Lee SO, Lin X, Trump DL, Gao AC (2006) Stat3 activation of NF-{kappa}B p100 processing involves CBP/p300-mediated acetylation. Proceedings of the National Academy of Sciences of the United States of America 103: 7264-7269.
- Dasgupta M, Unal H, Willard B, Yang J, Karnik SS, Stark GR (2014) Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. The Journal of biological chemistry 289: 30763-30771.
- Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. (2010) Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proceedings of the National Academy of Sciences of the United States of America 107: 21499-21504.
- Furtek SL, Backos DS, Matheson CJ, Reigan P (2016) Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS chemical biology 11: 308-318.
- Yue P, Turkson J (2009) Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs 18: 45-56. [Crossref]
- Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C, et al. (2012) Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood 119: 4614-4618.
- Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. (2015) The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signaling in rheumatoid arthritis. Annals of the rheumatic diseases 74: 1311-1316.
- Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, et al. (2014) The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. Journal of clinical pharmacology 54: 1354-1361.
- Abdelrahman RA, Begna KH, Al-Kali A, Hogan WJ, Litzow MR, et al. (2015) Momelotinib treatment-emergent neuropathy: prevalence, risk factors and outcome in 100 patients with myelofibrosis. Br J Haematol 169: 77-80. [Crossref]
- Namour F, Diderichsen PM, Cox E, Vayssiere B, Van der Aa A, Tasset C et al. (2015) Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection. Clinical pharmacokinetics 54: 859-874.
- Komrokji RS, Seymour JF, Roberts AW, Wadleigh M, To LB, Scherber R et al. (2015) Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood 125: 2649-2655.
- Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, Kim IS, et al. (2007) Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology 122: 607-614.
- Nam S, Wen W, Schroeder A, Herrmann A, Yu H, Cheng X, et al. (2013) Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Molecular oncology 7: 369-378.
- Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, et al. (2008) Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 112: 5095-5102.
- Schust J, Sperl B, Hollis A, Mayer TU, and Berg T (2006) Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chemistry and biology 13: 1235-1242.
- Arend RC, Londo Joshi AI, Gangrade A, Katre AA, Kurpad C, et al. (2016) Niclosamide and its analogs are potent inhibitors of Wnt/I²-catenin, mTOR and STAT3 signaling in ovarian cancer. Oncotarget 7: 86803-86815. [Crossref]
- Ma J, Xing W, Coffey G, Dresser K, Lu K, Guo A, et al. (2015) Cerdulatinib, a novel dual SYK/JAK kinase inhibitor, has broad anti-tumor activity in both ABC and GCB types of diffuse large B cell lymphoma. Oncotarget 6: 43881-43896.
- Fossey SL, Bear MD, Lin J, Li C, Schwartz EB, et al. (2011) The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 11: 112. [Crossref].
- Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. (2007) Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proceedings of the National Academy of Sciences of the United States of America 104: 7391-7396.
- Yang CL, Liu YY, Ma YG, Xue YX, Liu DG, Ren Y, et al. (2012) Curcumin blocks small cell lung cancer cells migration, invasion, angiogenesis, cell cycle and neoplasia through Janus kinase-STAT3 signalling pathway. PloS one 7: e37960.
- Zou M, Zhang X, Xu C (2016) IL6-induced metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of 3,3'-diindolylmethane in ovarian cancer cells. Cellular oncology 39: 47-57.
- Ai Y, Kang F, Huang Z, Xue X, Lai Y, et al. (2015) Synthesis of CDDO-amino acid-nitric oxide donor trihybrids as potential antitumor agents against both drug-sensitive and drug-resistant colon cancer. J Med Chem 58: 2452-2464. [Crossref].
- Verstovsek S, Hoffman R, Mascarenhas J, Soria JC, Bahleda R, McCoon P et al. (2015) A phase I, open-label, multi-center study of the JAK2 inhibitor AZD1480 in patients with myelofibrosis. Leukemia research 39: 157-163.