Abstract
Osteoprotegerin (OPG), a member of the tumor necrosis factor (TNF) receptor superfamily, suppresses osteoclast formation by preventing the binding of receptor activator of nuclear factor-kappa B ligand (RANKL) to receptor activator of NF-kappa B (RANK). Recent studies indicate that RANKL plays a critical role in the regulation of dendritic cells and macrophages. Indeed, RANK-RANKL interactions on these immune cells are involved in core host defense mechanisms against tumors and infection. Thus, OPG is thought to exert its effects by interfering with the RANKL-RANK interaction. While the role of OPG in bone metabolism are well described, little is known about its role in host defense. This review summarizes current knowledge about the role of OPG in pathologies such as cancer and microbial infections.
Key words
Osteoprotegerin (OPG), receptor activation of nuclear factor kappa B (RANK), RANK ligand (RANKL), TNF-related apoptosis-inducing ligand (TRAIL), host defense , infection disease
Introduction
Osteoprotegerin (OPG) is a member of the tumor necrosis factor (TNF) receptor superfamily, originally characterized according to its ability to suppress osteoclast formation by preventing the binding of receptor activator of nuclear factor-kappa B ligand (RANKL) to receptor activator of NF-kappa B (RANK) on osteoclast precursor cells. In 1997, two independent research groups identified OPG as a key regulator of bone turnover that functions by inhibiting osteoclastogenesis [1,2]. In the following year, Yasuda et al. reported that these independent findings actually pertained to the same molecule [3]. Since OPG-deficient mice also exhibit vascular calcification, OPG is thought to be a key link between bone and cardiovascular disease [4]. OPG is a secretory glycoprotein comprising 401 amino acids, 21 of which make up a signal peptide that is cleaved to generate the mature form (380 amino acids); this mature form contains seven domains: domains 1–4 are cysteine-rich N-terminal domains harboring motifs homologous to the TNF receptor and are necessary and sufficient for binding to RANKL, thereby inhibiting osteoclast differentiation and activity; domains 5 and 6 contain apoptosis-mediating death domain-homologous regions; and domain 7 is a C-terminal heparin-binding domain important for the interaction between OPG and proteoglycans. In addition, Cys 400 within domain 7 plays a central role in OPG dimer formation (Figure 1) [5].
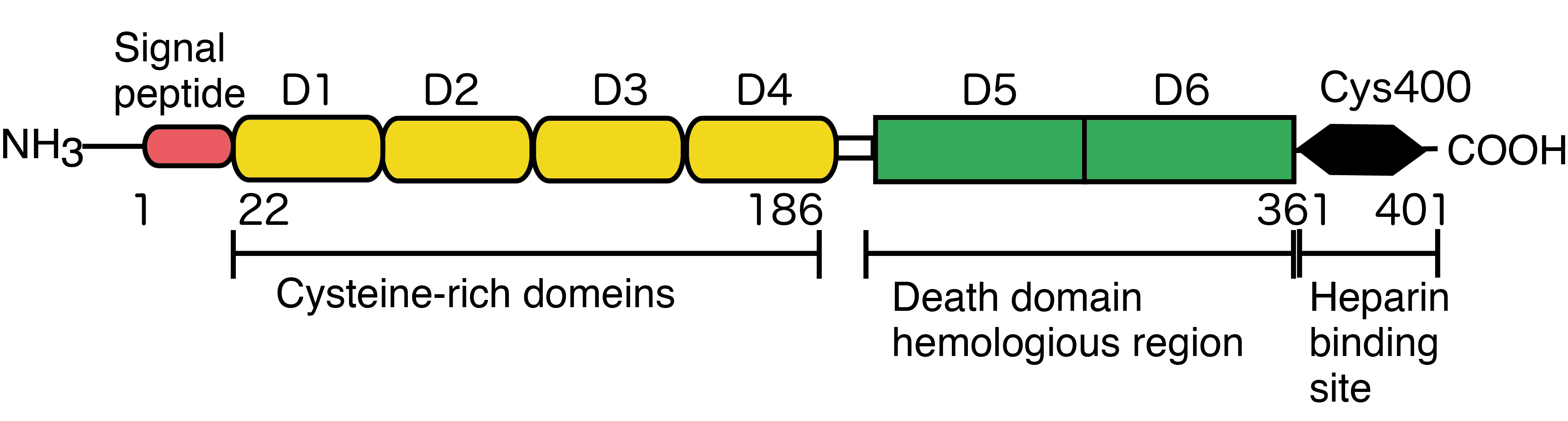
Figure 1. Molecular structure of OPG.
OPG is initially produced as a glycosylated monomer of 55–62 kDa, which then undergoes homodimerization before being secreted as a disulfide-linked homodimer (the mature 110–120 kDa form). The affinity of the OPG dimer for RANKL is much higher than that of the monomeric form [6]. OPG is expressed by many different cell types, including osteoclast precursors, mature osteoclasts, dendritic cells, B and T cells, fibroblasts, intestinal epithelial cells, vascular endothelial cells, and some cancer cells (e.g., breast and prostate cancers) [7]. The aim of this review is to provide an overview of the current knowledge about OPG-associated pathologies beyond bone remodeling, including cancer and microbial infections.
OPG prevents the RANKL-induced differentiation, fusion, and activation of osteoclasts
Osteoclasts are multinucleated bone resorbing cells formed by cytoplasmic fusion of their mononuclear precursors, which belong to the myeloid lineage of hematopoietic cells that also give rise to macrophages [8]. Upon binding RANKL, RANK (expressed by osteoclasts) activates six major signaling pathways: NFATc1, NF-kappa B, Akt/PKB, JNK, ERK, and p38, all of which play distinct roles in osteoclast differentiation, function, and survival [9-11]. OPG prevents the interaction between RANKL and RANK, thereby blocking osteoclast maturation [12].
OPG prevents TRAIL-induced apoptosis
In addition to regulating RANK-RANKL interactions during bone metabolism, OPG also stimulates cell survival by acting as a receptor for TNF-related apoptosis-inducing ligand (TRAIL) [13,14]. TRAIL is a member of the TNF superfamily, and induces apoptosis; it is secreted by normal tissues and preferentially induces apoptosis in tumor cells through the death domain receptors DR4 and DR5, both of which contain cytoplasmic death domains that activate apoptotic signaling pathways (Figure 2A) [15,16].
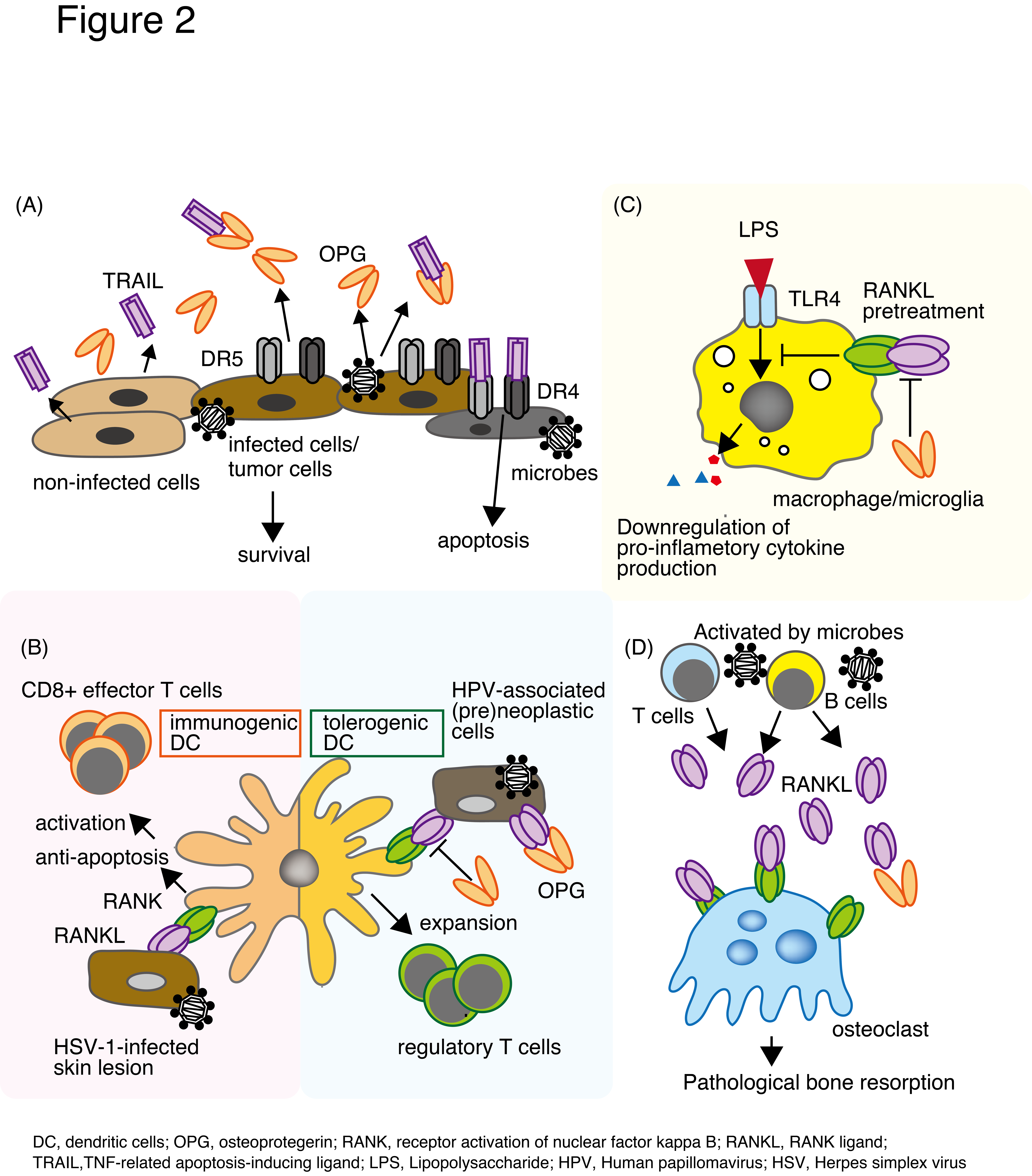
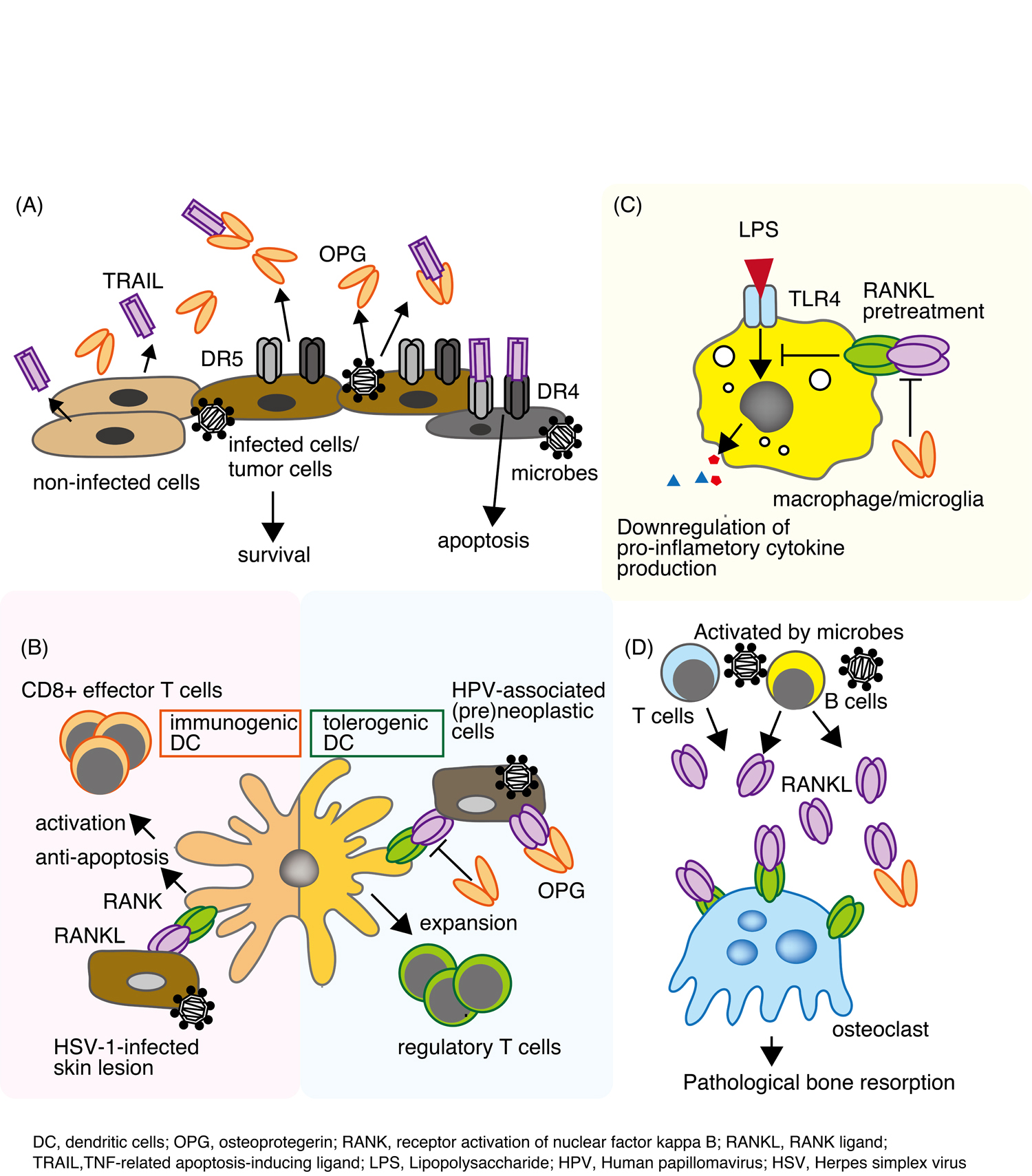
Figure 2. (A) TRAIL is secreted by normal tissues and preferentially promotes apoptosis of tumor cells or infected cells via death domain receptors (DR4 and/or DR5). Pathological cells produce OPG, which then blocks TRAIL-induced apoptosis. (B) RANKL-RANK signaling between HPV-associated (pre)neoplastic cells and tolerogenic DCs expands the regulatory T cell population. HSV-1-infected skin lesion prevent virus-induced DC apoptosis and (eventually) promote the emergence of CD8+ effector T cells. (C) RANKL suppresses the production of proinflammatory cytokines by macrophages in response LPS. (D) RANKL secreted by activated B cells and T cells promotes osteoclast maturity, leading to pathological bone resorption.
Results from clinical studies show that transformed endothelial cells that drive several types of tumors express higher levels of OPG than corresponding normal tissues; in breast cancer, this level of expression correlates with tumor grade [17-19]. In vitro studies show that OPG increases endothelial cell proliferation, migration, and angiogenesis through integrin alpha V beta 3 [20,21], and that breast cancer cells stimulate OPG production by endothelial cells through direct cell contact [22]. Several studies indicate that the tumor-promoting effects of OPG in breast cancer cells occur, at least in part, via a TRAIL-dependent pathway [18,23].
In addition to its ability to induce apoptosis in transformed tumor cells, TRAIL may also help to defend against microbial infections. Pathogenic microbes can induce TRAIL expression in both immune and non-immune cells and sensitize host cells to TRAIL-mediated apoptosis [24]. However, pathogenic microbes have evolved a variety of strategies to prevent TRAIL-mediated host cell death during the early stages of infection, which may enable them to replication and spread [25,26].
OPG regulates adaptive immune responses by controlling RANK-RANKL interactions on dendritic cells
RANKL and RANK were originally identified as a cytokine-receptor pair that controls the function of dendritic cells (DCs) [27,28]. DCs are professional antigen-presenting cells that are critical for inducing adaptive immunity and tolerance. DCs develop form myeloid- or lymphoid-committed progenitor cells [29,30]. RANKL binds to RANK on the surface of DCs, activating the NF-kappa B and JNK pathways and upregulating Bcl-xl expression, thereby initiating antiapoptotic signaling [31,32]. Recent studies demonstrate a crucial function of OPG during DC activation. DCs from OPG-deficient mice show better survival than DCs from wild-type mice; this is likely due to the absence of OPG, resulting in sustained RANK-RANKL interactions and enhanced DC survival and cytokine production in response to lipopolysaccharide (LPS) [33]. Accordingly, Yun et al., showed that DCs from OPG-deficient mice are more effective than wild-type DCs at stimulating allogeneic T cells [34]. An overview of the studies indicating a role for OPG in host defense against microbes is presented in Table 1
Table 1. Studies examining the association between osteoprotegerin and microbes (including LPS)
|
Author, date
|
Main finding
|
Related diseases
|
Source cells
|
Target cells
|
|
Chino T et al., 2009 [33]
|
DC from OPG-deficient mice survive better and produce more cytokines in response to LPS treatment than DC from wild-type mice.
|
Inflammatory response
|
ND
|
DCs
|
|
Tomlins C et al., 2010 [38]
|
E6 expression induces secretion of OPG and IL-6, which inhibit UV-induced apoptosis in HPV-negative cell lines and primary human keratinocytes.
|
HPV infection
|
Infected keratinocytes
|
HPV-negative keratinocytes
|
|
Demoulin SA et al., 2015 [39]
|
The cervical pre-neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells by RANK-RANKL interaction.
|
HPV infection
Cervical cancer
|
Infected cells/
pre-neoplastic cells
|
DCs
|
|
Klenner L et al., 2015 [45]
|
Cutaneous RANK-RANKL signaling upregulates CD8-mediated antiviral immunity during HSV infection.
|
HSV infection
|
Infected skin lesion
|
Langerhans cells
|
|
Castellanos-Gonzalez et al., 2008 [50]
|
OPG secreted from Cryptosporidium-infected intestinal epithelial cells blocks TRAIL-induced apoptosis.
|
Cryptosporidium infection
|
Infected intestinal epithelial cells
|
Infected intestinal epithelial cells
|
|
Maruyama K et al., 2006 [58]
|
Pretreatment with recombinant RANKL reduces the secretion of inflammatory cytokines by macrophages.
|
Endotoxic shock
Bacterial infection
|
ND
|
Macrophages
|
|
Shimamura M et al., 2014 [59]
|
RANKL exposure prevents LPS-triggered neuronal cell death.
|
Ischemic brain
|
ND
|
Microglias
|
|
Ashcroft AJ et al., 2003 [62]
|
T cell-mediated intestinal inflammation in IL-2-deficient mice is mitigated by OPG.
|
Inflammatory bowel disease
|
ND
|
DCs
|
|
Chen B et al., 2014 [69]
Kawai T et al., 2006 [70]
|
RANKL, secreted by B cells and T cells activated by periodontal pathogens, promotes osteoclast maturation, leading to alveolar bone resorption.
|
Periodontitis
|
B cells
T cells
|
Osteoclasts
|
DCs, dendritic cells; OPG, osteoprotegerin; RANK, receptor activation of nuclear factor kappa B; RANKL, RANK ligand; TRAIL,TNF-related apoptosis-inducing ligand;
LPS, lipopolysaccharide; HPV, human papillomavirus; HSV, herpes simplex virus; ND, not determined
Human papillomavirus infection
Human papillomavirus (HPVs) are small DNA viruses that specifically infect keratinocytes at different sites on the body. The prototypic skin cancer-associated HPV type 5 induces expression of the viral E6 oncoprotein [35], which is necessary for malignant transformation. E6 interferes with DNA damage responses and inhibits UV-induced apoptosis, suggesting that HPV type 5 contributes to early-stage tumorigenesis [36,37]. Tomoins et al. showed that E6 expression induces the secretion of OPG and interleukin (IL)-6, which inhibits UV-induced apoptosis in HPV-negative cell lines and primary human keratinocytes [38]. The authors concluded that increased HPV E6-induced secretion of both OPG and IL-6, but not either alone, may protect the infected host cell from UV-induced apoptosis via an autocrine mechanism, whereas a paracrine effect may protect neighboring non-HPV infected cells.
Furthermore, Demouline et al. demonstrated that HPV-associated cervical pre-neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells, which acquire the ability to promote the differentiation of naïve CD4+ T cells into regulatory T cells (Figure 2B) [39]. This phenomenon is caused by RANKL, which is secreted directly by cancer cells, and is abolished by OPG-mediated blockade of the RANKL/RANK signaling pathway. The authors suggested that the progression of genital HPV infections into pre-neoplastic lesions occurs because the antigens are not adequately recognized by the innate immune system, or not presented to the adaptive immune system. Expression of both RANKL and OPG is increased in pre-neoplastic cervical lesions; however, OPG expression remains stable during cervical cancer progression, suggesting that OPG expression in cervical cancer is probably not sufficient to counteract the neoplastic effects of RANKL expressed by DCs.
In addition, some reports present evidence that RANKL-mediated activation of T cells is associated with antimicrobial immunity [40-42]. For example, lymphocytic choriomeningitis virus and influenza virus can prime antigen-specific CD4+ T cells independently of the CD40 pathway, and induction of such responses in CD40-deficient mice is greatly inhibited by soluble RANK [41]. These findings suggest that, under specific circumstances, OPG may interfere with priming of CD4+ T cells caused by viral infections.
Viral infections and the OPG/RANKL/RANK system
Viral infections are controlled by the immune system, and CD8+ cytotoxic T-lymphocytes have are crucial for viral clearance [43,44]. Herpes simplex virus type 1 (HSV-1) is responsible for the majority of cutaneous viral infections. Klenner et al., demonstrated that cutaneous RANK-RANKL signaling improves the priming capacity of antiviral CD8+ effector T cells during HSV-1 infection of the skin [45]. They found that cutaneous RANK-RANKL signaling prevents virus-induced Langerhans cell apoptosis, improves antigen transport to regional lymph nodes, and increases the priming capacity of lymph node DCs for CD8+ cytotoxic T-lymphocytes by upregulating expression of toll-like receptor (TLR) 3 (Figure 2B). Importantly, injection of soluble RANKL protein into skin lesions protects mice from HSV-1 infection by inducing CD8+ antiviral effector T cells. Although the authors did not use recombinant RANK or OPG proteins to block RANKL signaling, these results suggest a potential role for OPG in HSV-1 infection.
Cryptosporidium infection
Cryptosporidium parasites are pathogens of human intestinal epithelial cells. To complete their life cycle, Cryptosporidium requires epithelial cells to survive for at least 48 h [46]. Data from studies of infected intestinal tissues suggest that apoptosis is increased in both infected and adjacent cells [46,47]. Initial invasion induces apoptosis of the infected cell; however, within a few hours, the parasite activates host cell NF-kappa B, which in turn activates an antiapoptotic mechanism [48, 49]. In vitro infection of human explants demonstrated upregulation of OPG in infected tissues [50]. These results suggested that OPG secreted from infected intestinal epithelial cells blocks TRAIL-induced apoptosis (Figure 2A). Interestingly, the authors also showed that the addition of recombinant OPG increased the infection rate to levels comparable with that observed in untreated cells. Furthermore, other studies suggest that pretreatment with OPG protects endothelial cells against detachment and apoptotic cell death induced by cysteine proteinases generated by Porphyromonas gingivalis, a major periodontal bacterium [51,52].
OPG and RANKL regulate proinflammatory cytokine production by macrophages
Macrophages are myeloid immune cells that reside in nearly all tissues; however, they display marked heterogeneity in terms of location, function, and cell surface marker expression. Activation of receptors such as TLRs or IL-1R on macrophages leads to activation of the I kappa B (IkB) kinase complex, resulting in its degradation [53,54]. IkB degradation promotes the release of free NF-kappa B, which then translocates to the nucleus and binds to the promoters of inflammatory genes to trigger their transcription. In addition to the “classical” NF-kappa B pathway, an “alternative” NF-kappa B signaling pathway has been described [55,56]. This signaling pathway is activated in response to developmental signals such as CD40 or RANK. Dejardin et al., suggested that crosstalk occurs between these pathways [57]. In addition, Maruyama et al., suggested that macrophage activation is regulated by the RANKL/RANK/OPG system via TLRs (Figure 2C) [58]. They also showed that pretreatment of macrophages with recombinant RANKL reduced their secretion of inflammatory cytokines in response to stimulation by bacteria and their components, such as LPS, flagellin, and CpGDNA [58]. Strikingly, prior administration of RANKL protected mice from LPS-induced death [58]. Other groups report that RANKL can prevent LPS-triggered neuronal cell death [59]. Also, a high serum OPG level is associated with an unfavorable outcome during ischemic stroke [60]. Enhanced RANKL/RANK signaling in OPG-deficient mice or recombinant RANKL-treated mice contributes to a reduction in infarction volume and brain edema by controlling post-ischemic inflammation [59]. In vitro, RANKL inhibits TLR4-mediated neuronal cell death in mixed cultures containing neurons and glial cells (Figure 2C) [59]. The molecular mechanisms by which RANKL affects TLR signaling in microglia remain to be clarified; however, RANKL treatment reduced expression of myeloid differentiation primary response 88 (MyD88) by bone marrow-derived macrophages in vitro [58]. Thus, RANKL might function downstream of TLR signaling in both microglia and macrophages. These data reveal therapeutic potential of RANKL for acute inflammatory diseases.
Inflammatory bowel disease
The role of RANKL signaling in the immune response has been studied using an IL-2 deficient mouse model, which is a model of spontaneous autoimmune disease [61]. Using this system, Ashcroft et al., reported that OPG affects gut inflammation, DC numbers, and macrophage activation in mice with inflammatory bowel and bone disease [62]. T cell-mediated intestinal inflammation in IL-2-deficient mice is significantly mitigated by administration of OPG, as is the decrease in the numbers of activated DCs in the intestine. By contrast, Stolina et al. showed that OPG overexpression does not cause obvious changes in immune responses [63]. Taken together, these results indicate that OPG may modulate inflammatory responses in the gut, but that such modulation may occur only under specific circumstances, or redundantly with other cytokine signals [64].
Periodontal disease
Periodontitis is an inflammatory disease characterized by periodontal pocket formation and alveolar bone resorption. Levels of RANKL mRNA are higher in cases of advanced periodontitis than in cases of moderate periodontitis or in healthy groups [65]. Excessive RANKL expression promotes the production of mature osteoclasts, which in turn leads to alveolar bone resorption. On the other hand, expression of OPG is associated with endothelial cells, and is significantly lower in periodontitis tissues than in healthy gingival tissue [66]. In addition, elevated RANKL levels are associated with the presence of P. gingivalis in clinical periodontal tissue samples [67]. RANKL and/or OPG are secreted by periodontal ligament fibroblasts, gingival fibroblasts, endothelial cells, and activated B and T cells [68,69]. Confocal microscopic analysis revealed that B and T cells are the cellular source of RANKL in bone resorptive lesion of periodontal disease [70] (Figure 2D).
During the early stages of periodontal disease, RANKL may be essential for the presence of macrophages and dendritic cells in periodontal tissue; these cells engulf and destroy pathogens. Apart from during osteoclastogenesis, our understanding of how the OPG/RANKL/RANK system regulates the response of periodontal tissue to periodontitis pathogens is rather limited. Further research is necessary to clarify the role of the OPG/RANKL/RANK system in the development of periodontitis.
OPG serum levels and infectious diseases
Epidemiologic studies suggest that serum OPG levels correlate with age, diabetes, hypertension, osteoporosis, and cardiovascular mortality [7]. OPG is expressed by arterial smooth muscle cells [71], in pulmonary artery smooth cells [72], and in the Weibel-Palade bodies (WPB) of endothelial cells in association with von-Willebrand factor (VWF) [73].
Some studies show that microbial infections affect the RANKL/OPG axis in the plasma. Human immunodeficiency virus type-1 (HIV-1), which causes chronic systemic inflammation, increased levels of proinflammatory cytokines, and helper T cell depletion, is associated with reduced expression of RANKL and a reduced RANKL/OPG ratio in the plasma [74]. In addition, HIV-infected patients have a significantly greater risk of cardiovascular disease and osteoporosis than non-infected individuals. Immune activation and immunosenescence of CD8+ T cells, together with OPG plasma levels, might be associated with the development and progression of early atherosclerosis [75]. Furthermore, a recent study examining a HIV transgenic rat model reported HIV-induced bone loss, whereby HIV infection leads to a decline in the frequency of B cells secreting OPG coupled with an increase in the frequency of B cells secreting RANKL [76]. These studies suggest that interfering with OPG may be a therapeutic option for infectious disease and chronic diseases such as cardiovascular disease or osteoporosis.
Djamiatun et al. showed that severe dengue hemorrhagic fever with thrombocytopenia, bleeding, and plasma leakage, is associated with exocytosis of WPB and increased circulating levels of VWF propeptide and OPG [77]. They suggested that the high OPG plasma levels observed in dengue fever may interfere with the antiviral and immune effects of TRAIL and with the RANK-RANKL system. On the other hand, an epidemiologic study reported that a single-nucleotide polymorphisms in the OPG gene are associated with an increased susceptibility to traveler’s diarrhea suffered by North American travelers to Mexico [78]. These studies suggest that OPG expression may correlate with susceptibility to infection. It remains to be established whether OPG is the cause or effect of infection.
Conclusion
OPG is mainly regarded as an antiresorptive cytokine that functions principally by binding to RANKL. In addition, accumulating evidence suggests that RANKL is important for immune responses; therefore, its role in host defense has received considerable attention. OPG is secreted by many types of cell in the context of several pathologies. In addition, OPG regulates the functions of dendritic cells, macrophages, and osteoclasts, which are highly specialized cells and closely related through the myeloid lineage. As described above, OPG is expected to have multifarious functions; however, the role played by this molecule in processes other than bone metabolism, in particular in relation to immune reactions, remain unclear. Therefore, further work is required to identify how OPG functions in host defense. Gaining insight into the various roles of OPG will help the development of new therapeutic strategies for inflammatory diseases, cancer, and microbial infections.
Acknowledgements
We would like to thank Prof. I. Holen of Department of Oncology, University of Sheffield for scientific discussion and critical reading of the manuscript.
2021 Copyright OAT. All rights reserv
References
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, et al. (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89: 309-319. [Crossref]
- Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, et al. (1997) Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun 234: 137-142. [Crossref]
- Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, et al. (1998) Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 139: 1329-1337. [Crossref]
- Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, et al. (1998) osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12: 1260-1268. [Crossref]
- Yamaguchi K, Kinosaki M, Goto M, Kobayashi F, Tsuda E, et al. (1998) Characterization of structural domains of human osteoclastogenesis inhibitory factor. J Biol Chem 273: 5117-5123. [Crossref]
- Schneeweis LA, Willard D, Milla ME (2005) Functional dissection of osteoprotegerin and its interaction with receptor activator of NF-kappaB ligand. J Biol Chem 280: 41155-41164. [Crossref]
- Pérez de Ciriza C, Lawrie A2, Varo N1 (2015) Osteoprotegerin in Cardiometabolic Disorders. Int J Endocrinol 2015: 564934. [Crossref]
- Udagawa N, Takahashi N, Yasuda H, Mizuno A, Itoh K, et al. (2000) Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 141: 3478-3484. [Crossref]
- Feng X (2005) Regulatory roles and molecular signaling of TNF family members in osteoclasts. Gene 350: 1-13. [Crossref]
- Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423: 337-342. [Crossref]
- Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, et al. (2002) RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature 416: 744-749. [Crossref]
- Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, et al. (2004) The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev 15: 457-475. [Crossref]
- Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, et al. (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273: 14363-14367. [Crossref]
- Baud'huin M, Duplomb L, Teletchea S, Lamoureux F, Ruiz-Velasco C, et al. (2013) Osteoprotegerin: multiple partners for multiple functions. Cytokine Growth Factor Rev 24: 401-409. [Crossref]
- Marsters SA, Sheridan JP, Pitti RM, Huang A, Skubatch M, et al. (1997) A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol 7: 1003-1006. [Crossref]
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, et al. (1997) Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277: 818-821. [Crossref]
- Holen I, Shipman CM (2006) Role of osteoprotegerin (OPG) in cancer. Clin Sci (Lond) 110: 279-291. [Crossref]
- Holen I, Cross SS, Neville-Webbe HL, Cross NA, Balasubramanian SP, et al. (2005) Osteoprotegerin (OPG) expression by breast cancer cells in vitro and breast tumours in vivo--a role in tumour cell survival? Breast Cancer Res Treat 92: 207-215. [Crossref]
- Cross SS, Yang Z, Brown NJ, Balasubramanian SP, Evans CA, et al. (2006) Osteoprotegerin (OPG)--a potential new role in the regulation of endothelial cell phenotype and tumour angiogenesis? Int J Cancer 118: 1901-1908. [Crossref]
- Kobayashi-Sakamoto M, Isogai E, Hirose K, Chiba I (2008) Role of alphav integrin in osteoprotegerin-induced endothelial cell migration and proliferation. Microvascular Research 76: 139-144.
- Kobayashi-Sakamoto M, Isogai E, Holen I (2010) Osteoprotegerin induces cytoskeletal reorganization and activates FAK, Src, and ERK signaling in endothelial cells. Eur J Haematol 85(1):26-35. [Crossref]
- Reid P1, Holen I (2009) Pathophysiological roles of osteoprotegerin (OPG). Eur J Cell Biol 88: 1-17. [Crossref]
- Holen I1, Croucher PI, Hamdy FC, Eaton CL (2002) Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res 62: 1619-1623. [Crossref]
- Sedger LM1, Shows DM, Blanton RA, Peschon JJ, Goodwin RG, et al. (1999) IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J Immunol 163: 920-926. [Crossref]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, et al. (1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386: 517-521. [Crossref]
- Peter ME1, Krammer PH (1998) Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol 10: 545-551. [Crossref]
- Wong BR, Rho J, Arron J, Robinson E, Orlinick J, et al. (1997) TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem 272: 25190-25194. [Crossref]
- Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, et al. (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390: 175-179. [Crossref]
- Chicha L, Jarrossay D, Manz MG (2004) Clonal type I interferon-producing and dendritic cell precursors are contained in both human lymphoid and myeloid progenitor populations. J Exp Med 200: 1519-1524. [Crossref]
- Manz MG, Traver D, Miyamoto T, Weissman IL, Akashi K (2001) Dendritic cell potentials of early lymphoid and myeloid progenitors. Blood 97: 3333-3341. [Crossref]
- Chen M, Wang YH, Wang Y, Huang L, Sandoval H, et al. (2006) Dendritic cell apoptosis in the maintenance of immune tolerance. Science 311: 1160-1164. [Crossref]
- Kriehuber E, Bauer W, Charbonnier AS, Winter D, Amatschek S, et al. (2005) Balance between NF-kappaB and JNK/AP-1 activity controls dendritic cell life and death. Blood 106: 175-183. [Crossref]
- Chino T, Draves KE, Clark EA (2009) Regulation of dendritic cell survival and cytokine production by osteoprotegerin. J Leukoc Biol 86: 933-940. [Crossref]
- Yun TJ, Tallquist MD, Aicher A, Rafferty KL, Marshall AJ, et al. (2001) Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J Immunol 166: 1482-1491. [Crossref]
- Hebner CM, Laimins LA (2006) Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol 16: 83-97. [Crossref]
- Garnett TO, Duerksen-Hughes PJ (2006) Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Arch Virol 151: 2321-2335. [Crossref]
- Struijk L, van der Meijden E, Kazem S, ter Schegget J, de Gruijl FR, et al. (2008) Specific betapapillomaviruses associated with squamous cell carcinoma of the skin inhibit UVB-induced apoptosis of primary human keratinocytes. J Gen Virol 89: 2303-2314. [Crossref]
- Tomlins C1, Storey A (2010) Cutaneous HPV5 E6 causes increased expression of Osteoprotegerin and Interleukin 6 which contribute to evasion of UV-induced apoptosis. Carcinogenesis 31: 2155-2164. [Crossref]
- Demoulin SA, Somja J, Duray A, Guenin S, Roncarati P, et al. (2015) Cervical (pre)neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells via RANKL secretion. Oncoimmunology 4: e1008334. [Crossref]
- Miyahira Y, Akiba H, Katae M, Kubota K, Kobayashi S, et al. (2003) Cutting edge: a potent adjuvant effect of ligand to receptor activator of NF-kappa B gene for inducing antigen-specific CD8+ T cell response by DNA and viral vector vaccination. J Immunol 171(12): 6344-6348. [Crossref]
- Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, et al. (1999) TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med 189: 1025-1031. [Crossref]
- Padigel UM, Kim N, Choi Y, Farrell JP (2003) TRANCE-RANK costimulation is required for IL-12 production and the initiation of a Th1-type response to Leishmania major infection in CD40L-deficient mice. J Immunol 171: 5437-5441. [Crossref]
- van Lint A, Ayers M, Brooks AG, Coles RM, Heath WR, et al. (2004) Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol 172: 392-397. [Crossref]
- Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, et al. (2013) Immune surveillance by CD8αα+ skin-resident T cells in human herpes virus infection. Nature 497: 494-497. [Crossref]
- Klenner L, Hafezi W, Clausen BE, Lorentzen EU, Luger TA, et al. (2015) Cutaneous RANK-RANKL Signaling Upregulates CD8-Mediated Antiviral Immunity during Herpes simplex Virus Infection by Preventing Virus-Induced Langerhans Cell Apoptosis. J Invest Dermatol. [Crossref]
- Chen XM, Levine SA, Tietz P, Krueger E, McNiven MA, et al. (1998) Cryptosporidium parvum is cytopathic for cultured human biliary epithelia via an apoptotic mechanism. Hepatology 28: 906-913. [Crossref]
- McCole DF, Eckmann L, Laurent F, Kagnoff MF (2000) Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect Immun 68: 1710-1713. [Crossref]
- Widmer G, Corey EA, Stein B, Griffiths JK, Tzipori S (2000) Host cell apoptosis impairs Cryptosporidium parvum development in vitro. J Parasitol 86: 922-928. [Crossref]
- Ojcius DM, Perfettini JL, Bonnin A, Laurent F (1999) Caspase-dependent apoptosis during infection with Cryptosporidium parvum. Microbes Infect 1: 1163-1168. [Crossref]
- Castellanos-Gonzalez A, Yancey LS, Wang HC, Pantenburg B, Liscum KR, et al. (2008) Cryptosporidium infection of human intestinal epithelial cells increases expression of osteoprotegerin: a novel mechanism for evasion of host defenses. J Infect Dis 197: 916-923. [Crossref]
- Kobayashi-Sakamoto M, Hirose K, Nishikata M, Isogai E, Chiba I (2006) Osteoprotegerin protects endothelial cells against apoptotic cell death induced by Porphyromonas gingivalis cysteine proteinases. FEMS Microbiol Lett 264: 238-245. [Crossref]
- 52. Kobayashi-Sakamoto M, Hirose K, Isogai E, Chiba I (2004) NF-kappaB-dependent induction of osteoprotegerin by Porphyromonas gingivalis in endothelial cells. Biochemical and biophysical research communications 315: 107-112. [Crossref]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, et al. (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25: 677-686. [Crossref]
- Biswas SK, Lewis CE (2010) NF-κB as a central regulator of macrophage function in tumors. J Leukoc Biol 88: 877-884. [Crossref]
- Vallabhapurapu S, Karin M (2009) Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27: 693-733. [Crossref]
- Bonizzi G, Karin M (2004) The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25: 280-288. [Crossref]
- Dejardin E (2006) The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 72: 1161-1179. [Crossref]
- Maruyama K, Takada Y, Ray N, Kishimoto Y, Penninger JM, et al. (2006) Receptor activator of NF-kappa B ligand and osteoprotegerin regulate proinflammatory cytokine production in mice. J Immunol 177: 3799-3805. [Crossref]
- Shimamura M, Nakagami H, Osako MK, Kurinami H, Koriyama H, et al. (2014) OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc Natl Acad Sci U S A 111: 8191-8196. [Crossref]
- Song TJ, Kim J, Yang SH, Park JH, Lee HS, et al. (2012) Association of plasma osteoprotegerin levels with stroke severity and functional outcome in acute ischaemic stroke patients. Biomarkers 17: 738-744. [Crossref]
- Schimpl A, Berberich I, Kneitz B, Krämer S, Santner-Nanan B, et al. (2002) IL-2 and autoimmune disease. Cytokine Growth Factor Rev 13: 369-378. [Crossref]
- Ashcroft AJ, Cruickshank SM, Croucher PI, Perry MJ, Rollinson S, et al. (2003) Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity 19: 849-861. [Crossref]
- Stolina M, Dwyer D, Ominsky MS, Corbin T, Van G, et al. (2007) Continuous RANKL inhibition in osteoprotegerin transgenic mice and rats suppresses bone resorption without impairing lymphorganogenesis or functional immune responses. J Immunol 179: 7497-7505. [Crossref]
- Akiyama T, Shinzawa M, Akiyama N (2012) RANKL-RANK interaction in immune regulatory systems. World J Orthop 3: 142-150. [Crossref]
- Liu D, Xu JK, Figliomeni L, Huang L, Pavlos NJ, et al. (2003) Expression of RANKL and OPG mRNA in periodontal disease: possible involvement in bone destruction. Int J Mol Med 11: 17-21. [Crossref]
- Crotti T, Smith MD, Hirsch R, Soukoulis S, Weedon H, et al. (2003) Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J Periodontal Res 38: 380-387. [Crossref]
- Wara-aswapati N, Surarit R, Chayasadom A, Boch JA, Pitiphat W (2007) RANKL upregulation associated with periodontitis and Porphyromonas gingivalis. J Periodontol 78: 1062-1069. [Crossref]
- Kajiya M, Giro G, Taubman MA, Han X, Mayer MP, et al. (2010) Role of periodontal pathogenic bacteria in RANKL-mediated bone destruction in periodontal disease. J Oral Microbiol 2. [Crossref]
- Chen B, Wu W, Sun W, Zhang Q, Yan F, et al. (2014) RANKL expression in periodontal disease: where does RANKL come from? Biomed Res Int 2014: 731039. [Crossref]
- Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, et al. (2006) B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol 169: 987-998. [Crossref]
- Schoppet M, Kavurma MM, Hofbauer LC, Shanahan CM (2011) Crystallizing nanoparticles derived from vascular smooth muscle cells contain the calcification inhibitor osteoprotegerin. Biochem Biophys Res Commun 407: 103-107. [Crossref]
- Lawrie A, Waterman E, Southwood M, Evans D, Suntharalingam J, et al. (2008) Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am J Pathol 172: 256-264. [Crossref]
- Zannettino AC, Holding CA, Diamond P, Atkins GJ, Kostakis P, et al. (2005) Osteoprotegerin (OPG) is localized to the Weibel-Palade bodies of human vascular endothelial cells and is physically associated with von Willebrand factor. J Cell Physiol 204: 714-723. [Crossref]
- Kelesidis T, Kendall MA, Yang OO, Hodis H, Currier JS (2013) Perturbations of circulating levels of RANKL-osteoprotegerin axis in relation to lipids and progression of atherosclerosis in HIV-infected and -uninfected adults: ACTG NWCS 332/A5078 Study. AIDS Res Hum Retroviruses 29: 938-948. [Crossref]
- D'Abramo A, Zingaropoli MA, Oliva A, D'Agostino C, Al Moghazi S, et al. (2014) Immune activation, immunosenescence, and osteoprotegerin as markers of endothelial dysfunction in subclinical HIV-associated atherosclerosis. Mediators Inflamm 2014: 192594. [Crossref]
- Titanji K, Vunnava A, Sheth AN, Delille C, Lennox JL, et al. (2014) Dysregulated B cell expression of RANKL and OPG correlates with loss of bone mineral density in HIV infection. PLoS Pathog 10: e1004497. [Crossref]
- Djamiatun K, van der Ven AJ, de Groot PG, Faradz SM, Hapsari D, et al. (2012) Severe dengue is associated with consumption of von Willebrand factor and its cleaving enzyme ADAMTS-13. PLoS Negl Trop Dis 6: e1628. [Crossref]
- Mohamed JA, DuPont HL, Jiang ZD, Flores J, Carlin LG, et al. (2009) A single-nucleotide polymorphism in the gene encoding osteoprotegerin, an anti-inflammatory protein produced in response to infection with diarrheagenic Escherichia coli, is associated with an increased risk of nonsecretory bacterial diarrhea in North American travelers to Mexico. J Infect Dis 199: 477-485. [Crossref]