Abstract
The cadherin/catenin complex regulates diverse signaling pathways through cell-cell interaction in development, differentiation, and ischemia and neurodegenerative disorders such as Alzheimer’s disease (AD). Several stimuli affecting cadherin-mediated signaling regulate the processing of membrane-spanning proteins, including amyloid precursor protein (APP), downregulate wnt-related signaling, or promote nuclear transcriptional program. Nuclear phosphoinositide signaling regulates stress-mediated chromatin modification and contributes to the development of cell polarity or malignancy in cancers. Interestingly, the vascular endothelial (VE)-cadherin/catenin complex can modulate the endothelial barrier function and blood vessel stability under inflammatory conditions provoked by hypoxia or stroke. Blood-brain barrier (BBB) permeability and the availability of nutrients are prerequisite for normal brain functions. Occurrence of stroke, hypoxia-ischemia encephalopathy disrupts the homeostasis regulated by VE-cadherin/vascular endothelial growth factor (VEGF) receptor signaling, a process that differs between adult and neonatal brains. N-cadherin and VEGF regulate the angiogenesis or vascular response and recruitment or migration of neural progenitor cells to demyelinated lesions. The hypoxia-inducible factor (HIF) signaling axis activates key molecules such as netrin-1 and wnt-mediated catenin signaling in endothelial cells and oligodendrocyte precursor cells (OPCs) in the subventricular zone (SVZ) or subcortical white matter tract, and spatiotemporal dysregulation of HIF signaling may be the primary cause of periventricular leukomalacia. In this review, we discuss the cadherin/catenin-mediated machinery and relevant associated pathological disorders, focusing on the organization of the complicated molecular framework as well as on several developmental functions mediated by cadherin/catenin complex.
Key words
cadherin, hypoxia, p120 catenin, periventricular leukomalacia, presenilin
Introduction
Cadherin/catenin cell adhesion complexes are necessary for synaptogenesis, plasticity, endothelial survival, and vascular morphogenesis [1,2]. These cell adhesion machineries are regulated by several catenins, including a-, b-, and d-, or p120 catenins possessing armadillo (Arm) repeats in their central domains. a-Catenin interacts with several actin-binding proteins such as a-actinin and modulates the dynamics of actin cytoskeletons. Its interacting partner, b-catenin, constitutes the cadherin-mediated adherens junctions through the interaction with classical E- or N-cadherins [3]. b-Catenin also regulates synaptic homeostasis through binding to PDZ-domain-containing proteins, and modulates the neuronal activity or dendritic morphogenesis in neurons [4]. p120 catenin binds classical cadherins through their juxtamembrane (JMD) sequences and regulates their processing by presenilin-1 (PS1) [5,6]. Both p120 catenins and the armadillo repeat protein deleted in velo-cardio-facial syndrome (ARVCF) subfamily can exist as two major splicing isoforms that differ by the presence (isoform 1) or absence (isoform 3) of a coiled-coil region at the N-terminus, and regulate the classical cadherin transport and stability in the plasma membrane [7]. Interestingly, delta-interacting protein A (DIPA) was identified as an isoform 1-specific p120 binding protein. Both knockdown and overexpression of DIPA cause phenotypes similar to those seen in N-cadherin mutants, referred to hydrocephalus and heterotopia, which are implicated in aberrant cadherin-mediated signaling during brain development [8]. Furthermore, p120 catenin is involved in PS1-mediated cadherin processing, which could compete with APP cleavage organized by the g-secretase complex in a context-dependent manner [6]. In addition, nuclear phosphatidylinositol-4,5-bisphosphate PI(4,5)P2-mediated signaling is necessary for the regulation of transcription and promotes E-cadherin biogenesis or suppresses inhibitor of growth protein 2 (ING2)-mediated chromatin remodeling in response to several stimuli [9].
Under hypoxic conditions, HIF-1 is stabilized by suppression of proteasomal degradation of its subunit HIF-1a, and both PS1 and PS2 are known to regulate the HIF-1a turnover [10]. PS1 and PS2 promote hypoxia-dependent HIF-1a expression, which is dependent on APP or its intracellular domain (AICD) generation by g-secretase. On the other hand, HIF-1 signaling is responsible for the upregulation of vascular endothelial growth factor (VEGF) detected in cerebral infarction after middle cerebral artery occlusion (MCAO) or neonatal stroke, and VE-cadherin is required for VEGF-mediated angiogenesis [1,11]. VEGF controls endothelial permeability by stimulating the b-arrestin-dependent endocytosis of VE-cadherin [12]. Interestingly, disruption of BBB function after ischemic stroke differs among neonates and adults. In fact, expression of tight junction proteins is retained to a great extent in the immature brain than in the adult brain after the stroke, and several genes that increase vascular permeability, such as VEGF receptor 2, are overexpressed only in the adult brain [13,14].
Glial fibrillary acidic protein (GFAP)-positive astrocytic cells from the SVZ can give rise to neurons, NG2-positive oligodendrocyte progenitor cells (OPCs), and mature myelinating oligodendrocytes [15]. OPC-intrinsic HIF1/2a is required for postnatal myelination by coupling with white matter angiogenesis as shown by loss of white matter tracts in forebrain of the conditional HIF1/2a knockout mouse, which phenotype is reminiscent of periventricular leukomalacia (PVL) categorized as the disorder in preterm infants [16]. According to a model of subcortical white matter demyelination, expansion of neuronal progenitor cells (NPCs) in the SVZ, and activation of N-cadherin signaling by a disintegrin and metalloproteinase (ADAM) protease in NPCs are both necessary for enhanced migration of NPCs into demyelinated lesions [17]. OPC expansion due to white matter injury caused by hypoxic-ischemia and death of SVZ progenitors in the recovery phase seems to be common features in hypoxic-ischemic encephalopathy (HIE) or neonatal hypoxia [17-20]. In this review, we especially focus on recently unveiled signaling pathway for cadherin-dependent cell/cell communication during the developmental disorders and neonatal hypoxia, including PVL.
Molecular organization of cadherin/catenin complex
Cadherin family proteins are divided into several subgroups based on their structure and function. Classical cadherins are type I transmembrane proteins with an extracellular domain containing five EC subdomains (EC1–EC5), which mediate stabilization for homophilic interactions via Ca2+ binding [2]. Their cytoplasmic domain contains the juxtamembrane (JMD) sequence required for binding to p120 catenin and interacts with b-catenin at the C-terminal region. b-Catenin regulates the actin cytoskeleton through interaction with a-catenin, and EPLIN (epithelial protein lost in neoplasm) and Afadin mediates the linkage between F-actin and cadherin/catenin complex [21,22]. The p120 catenin family consists of p120 catenin, ARVCF, d-catenin/NPRAP, plakophilins, and p0071. Both p120 catenin and ARVCF exist prevalently as two splicing variants with or without an N-terminal regulatory coiled-coil domain (isoforms 1 and 3). ARVCF, d-catenin, and p0071 have partial overlapping functions in synapses or spines, and possess a PDZ domain at the C-terminal domain [7]. d-Catenin knockdown affects the spine and synaptic density independently of cadherin binding, suggesting the biological importance of the PDZ domain. p120 catenin has coordinated function between cadherin and Rho family proteins and regulates spine and synaptic morphogenesis [23]. p120 catenin also plays a critical role in cadherin processing and competes with APP processing, which is mediated by limited amounts of g-secretase complex containing presenilin-1 (PS1) [6].
p120 isoform 1 (p120-1), containing an N-terminal coiled-coil domain generated by splicing, is mainly expressed in mesenchymal lineages or neurons (Figure 1). In contrast, p120 isoform 3 (p120-3) is predominantly expressed in epithelial cells. The epithelial-to-mesenchymal transformation (EMT) is coordinated with p120-3 or 4 to p120-1 alternative switching and classical E- to N-cadherin transition [24]. DIPA family proteins specifically bind p120-1, but not the p120-3, and colocalize with adherens junctions. Interestingly, knockdown or overexpression of DIPA causes a defect in neural tube closure and neuroepithelial disorganization similar that seen in a model of N-cadherin mutation in zebrafish [8]. Coiled-coil domain containing protein 85C (Ccdc85c), a DIPA family protein, is known to be disrupted in a hemorrhagic hydrocephalus (hhy) mutant mouse model [25]. Since Ccdc85c is recruited to the adherens junction via p120-1, it is suggested that a defect in Ccdc85c/p120 association causes subcortical heterotopia and hydrocephaly.
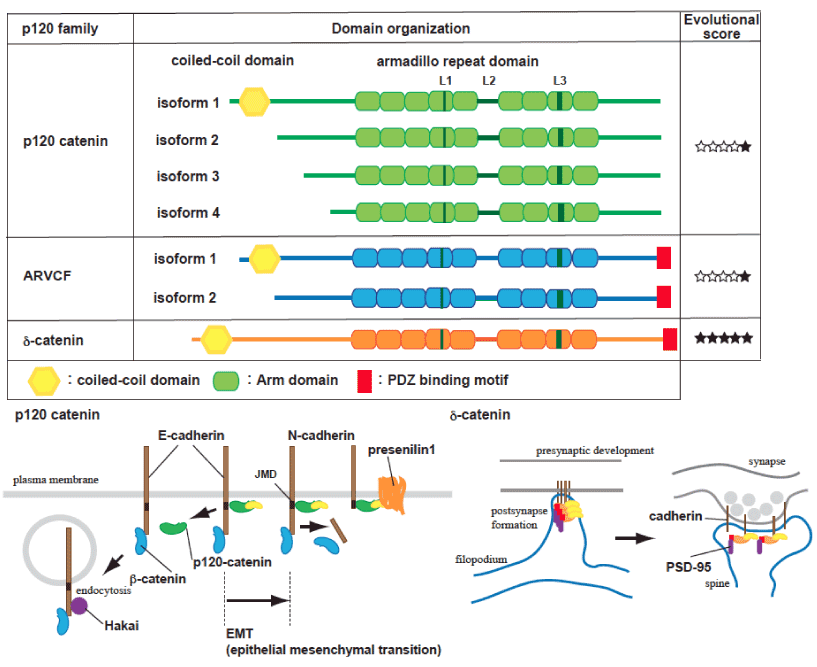
Figure 1. Schematic representation of p120 catenin family proteins.
p120 catenin gene contains four different translation initiation sites (isoform 1-4) and multiple protein variants are generated by alternative splicing occurring in the N-terminus and in the C-terminus. Most cells express multiple forms of p120 catenin, all possessing 9 central tandem repeats of Armadillo domains with L domain (L1-L3) and alternatively spliced exons in the middle and C-terminal regions (A-C). p120 catenin isoform 1 contains a coiled-coil domain and multiple phosphorylated regions at N-terminus. p120 catenin isoform 3 and 4, which lack the coiled-coil domain and most of N-terminal regulatory domain, are mainly expressed in epithelial cells. The N-terminal regulatory domain has phosphorylation sites and is necessary for receptor tyrosine kinase-induced cell motility in response to epidermal growth factor and hepatocyte growth factor. ARVCF has two major isoforms like p120 catenin, and ARVCF and d-catenin contain a PDZ domain at the C-terminal tail, which mediates the interaction with PSD-95 [47]. Filled star symbols indicate the presence of an orthologue of the corresponding p120 catenin family protein in phylogenetic history, denoted as Ctenophores, Cnidarians, Protostomes, Chordates, and Vertebrates (7). Cadherin complexes are regulated under a dynamic equilibrium at the plasma membrane, dependent on association with p120 catenin family proteins. PS1 is also required for the cadherin turnover, through restrictive proteolysis in the balance of APP processing [6]. Conversion of N- to E-cadherin occurs during the epithelial-to-mesenchymal transformation (EMT), and p120 catenin-unbound cadherin is targeted to internalization and subjected to Hakai-mediated degradation [28, 30, 32]. a- and b-catenin have important roles in clustering cadherins and regulating cytoskeletal dynamics, but their effects on the surface cadherin levels are considered to be indirect. d-catenin, which can be localized in dendrites or synapses, is involved in the regulation of PSD95 and N-cadherin levels, Interaction with the PDZ domain binding protein, but not cadherin binding, is necessary for activity-dependent d-catenin-mediated signal [48].
Cadherin/catenin function in pathological condition
Ependymal precursors generate radial glia on the ventricular wall surface of the fetus and ciliated ependymal cells at birth; however, hhy mutant brains exhibit ependymal agenesis with inflammatory degeneration [25]. Disruption of N-cadherin-based adherens junction leads to ependymal apoptosis and denudation of brain ventricular walls [26]. During brain development, p120 catenin and N-cadherin colocalize in neuroblasts in proliferative SVZ. The defects in neuroblast migration caused by the dysfunction of molecular machinery involving E-cadherin/b-catenin can be detected in hyh mutants (hydocephalus with hop gait) carrying missense mutations in Napa, which encodes soluble N-ethylmaleimide sensitive factor attachment protein alpha (aSnap) and affects organization of the ventricular neuroepithelium [27].
p120 catenin is involved in the stabilization of classical cadherins at the plasma membranes by preventing endocytosis, and substantial loss of p120 catenin causes loss of E-cadherin during cancer progression to a metastatic stage [28]. In contrast, Numb binds the p120/E-cadherin complex by directly interacting with the NVYYY motif and promotes E-cadherin endocytosis [29]. The motif in E-cadherin is phosphorylated by Src kinase and recruits a c-Cbl-related E3 ubiquitin ligase, Hakai, which results in endocytosis and degradation [30-32]. On the other hand, E-cadherin biogenesis is regulated by several signaling mechanism related to intrinsic or exogenous stimuli [9,33]. The vitamin D receptor ligand, 1a,25-dihydroxyvitamin D3 (1a,25(OH)2D3), activates RhoA and p38 mitogen-activated protein kinase (p38 MAPK) accompanied by Ca2+ influx, resulting in the promotion of differentiation with E-cadherin expression [33]. In contrast to the Ca2+-induced mechanism, nuclear PI(4,5)P2 generation catalyzed by phosphatidylinositol-5-phosphate 4-kinase type II beta (PIPKIIb) was found to be critical for E-cadherin upregulation in colon cancer model, after treatment with the vitamin D receptor ligand, 1a,25-dihydroxyvitamin D3 (1a,25(OH)2D3) [9] (Figure 2). Phosphoinositide 5’-phosphate (PI(5)P) interacts with ING2, a tumor suppressor protein containing a plant homeodomain finger motif very common in chromatin-regulatory proteins and ING2 promotes stress-induced cell death via the p53 activation pathway [34]. ING2 comprises part of a chromatin-regulatory complex through direct association with PI(5)P, in response to genotoxic stress. This ING2/PI(5)P interaction is a prerequisite for stabilization of promoter occupancy, allowing regulation of cell cycle progression [35]. These results suggest that nuclear PI(4,5)P2 and PI(5)P might counteract to control the cell growth and adhesive properties through sensing several outside signals.
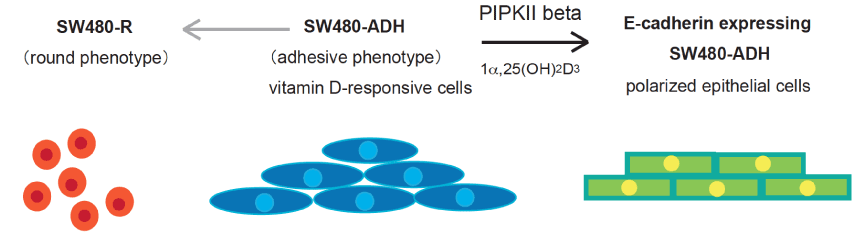
Figure 2. Epithelial-like polarization of SW480 cells by PI(5)P 4-kinase IIb-mediated nuclear PI(4,5)P2 signaling.
SW480 cells are a model of human colorectal carcinoma and utilized to study phenotypic changes of tumor progression and the epithelial-like polarization model [49]. SW480R cells (left) lacking Vitamin D receptor expression show rounded morphology and fast anchorage-independent growth, with loss of contact inhibition and adhesion. In contrast, SW480-ADH cells (middle) retains properties of ligand (1a,25(OH)2D3)-activated VDR activation and acquire the epithelial morphology with E-cadherin expression. Phosphatidylinositol 5-phosphate 4-kinase type II beta (PI(5)P 4-kinase IIb) mediates nuclear PI(4,5)P2 generation upon VDR activation and is required for transcriptional E-cadherin upregulation in SW480-ADH cells [9].
Angiopoietin-like 4 (Angtpl4) is induced by hypoxia and exhibits a proangiogenic response independent of VEGF [36]. Angtpl4 induces significantly decrease in infarct region in ischemic stroke model presumably due to retained VE-cadherin integrity and increased PECAM stability protecting blood vessel integrity. miR-27a was identified as specifically targeting VE-cadherin, and downregulation of miR-27a is known to be required for angiogenesis consistent with the reduction of vascular permeability in response to VEGF by miR-27 inhibitor, suggesting the importance of hypoxia-dependent regulation of VE-cadherin/VEGF signaling by transcriptional regulation [37].
Cadherin-mediated signaling in hypoxic/ischemic response
HIF-1 and HIF-2 are transcriptional heterodimers composed of an O2-regulated HIF-1a or HIF-2a subunit and a constitutively stable HIF-1b [38]. In the presence of O2, both HIF-a subunits are subjected to hydroxylation by prolyl-4-hydroxylase domain (PHD) proteins, which results in their proteasomal degradation. However, during hypoxic conditions, the hydroxylation process is suppressed and HIFs form stable complexes that activate transcription of hypoxia-responsive genes such as VEGF or BNIP3. Hypoxia-dependent HIF-1a expression is promoted by both PS1 and PS2, as well as tightly regulated PHD2 expression. HIF-1a induction is dependent on APP or AICD generation by g-secreatse but not on the Notch intracellular domain (NICD) -mediated transactivation [10]. Interestingly, development of oligodendrocyte precursor cells (OPCs) requires extraordinary nutritive demands for wrapping numerous myelin segments to axons, and HIF1/2a activation tightly regulates wnt7a/b-mediated b-catenin signaling in vessel growth into developing white matter tracts [16].
VEGF is the main regulator of angiogenesis and blood vessels localized in adult SVZ regulate neurogenesis and neuroblast migration as vascular niche [39]. It has been reported that SVZ neuroblasts exhibit proliferative response to ischemic stress concomitant with loss of migrative property in middle cerebral artery occlusion (MCAO) model [40]. Ependymal cells after stroke induce glial fibrillary acidic proteins (GFAP) and vascular network. Expression of VEGF and VEGF receptors is upregulated in the ischemic border region with invading vessel growth by permanent occlusion of the middle cerebral artery [11]. Prolonged intraventricular VEGF supply promotes angiogenesis and pericyte coverage of brain endothelial cells with increase in cerebral blood flow via upregulation of N-cadherin on cerebral microvessels during focal cerebral ischemia [41].
A specific VEGF-dependent period of angiogenesis is important for the development and maintenance of the periventricular vessels and functional BBB, as was shown by expressing inducible secreted VEGF decoy receptors in the brain of transgenic mice [42]. An anti-VEGF therapy against vascular dysfunction at the acute phase of ischemia has been proposed to effectively block edema formation and tissue damage without change in regional blood flow [43]. Interestingly, significant delay in angiogenesis has been identified after stroke in a neonatal model of hypoxia-induced encephalopathy as was shown by no stimulation of endothelial proliferation in peri-ischemic regions after stroke [44]. Furthermore, increased vascular BBB and inhibition of neutrophil transmigration after stroke have been shown in neonates, and the different response is attributed to the preserved molecular components on the basal lamina components and to adhesion molecules such as laminin and zonula occludens protein 1 in injured regions of the neonates [13].
In a demyelination model, dietary cuprizone-fed mice exhibited chronic demyelination in subcortical white matter, while lysolecithin treatment induced acute focal demyelination in the corpus callosum [17]. In both animal models, upregulation of ADAM10 and N-cadherin, together with their binding proteins b-catenin and p120 catenin, were detected in quiescent GFAP-positive neural stem cells and EGFR-positive amplifying NPCs during the peak period. Migrating EGFR-positive NPCs from the SVZ niche possess high N-cadherin processing activity and are recruited to the lesion in an ADAM10-dependent manner. GFAP-positive primary progenitor (type B) cells, which penetrate the ependymal layer, also generate NG2-positive OPCs and myelinating oligodendrocytes. In demyelinating lesions, migrating NG2+ cells are significantly increased with higher EGFR expression than cortical nonmigrating NG2+ cells [15,20]. The pathological response of progenitor recruitment seems to require the physical association with vascular reactivity, and both netrin-1 and VEGF have been identified as the key molecules in this process [19]. A similar underlying mechanism could regulate pathological processes in periventricular leukomalacia [45]. Embyonic VEGF blockade affects neurons migrating after damage in lateral ganglionic eminence but does not affect lamination of excitatory pyramidal neurons. This suggests that primary periventricular lesions caused by loss of periventricular vessels induces secondary neuropathology like the PVL phenotype [42, 46].
Conclusions
The cadherin/catenin system is modulated by numerous interacting proteins. The responses of this system to environmental stimuli control synaptic function, and vascular development and homeostasis, also establishing cell polarity through cell/cell communication. Expression and stability of cadherins are modulated by nuclear lipid signaling-mediated PI(4,5)P2 and miRNA-mediated signaling in several pathological conditions. p120 catenins modulate several processes, including PS1-mediated APP processing through cadherin binding and proteolysis, and migration of neural progenitors in ischemic injury. HIFs mediates several hypoxic responses, including VEGF-mediated angiogenesis during brain development. Several N- or VE-cadherin and basal lamina component-mediated signaling processes are required for repair processes of neonatal hypoxia or periventricular pathogenesis. The molecular pathways seems to be different from adult pathogenesis, and elucidating the underlying mechanisms controlled by cadherins can create therapeutic tools to circumvent neuronal death and hypomyelination in neonates.
Acknowledgement
I thank Dr. Robakis NK at Mount Sinai School of Medicine in New York for permission to mention the summary of our collaborative work.
Reference
- Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, et al. (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98: 147-157. [Crossref]
- Ouyang M, Lu S, Kim T, Chen CE, Seong J, et al. (2013) N-cadherin Regulates Spatially Polarized Signals through Distinct β-Catenin-dependent P120ctn and beta-Catenin Signaling Pathways. Nat Commun 4: 1589.
- Fannon AM, Colman DR (1996) A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17: 423-434. [Crossref]
- Yu X, Malenka RC (2003) Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 6: 1169-1177. [Crossref]
- Piedra J, Miravet S, Castaño J, Pálmer HG, Heisterkamp N, et al. (2003) p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin interaction. Mol Cell Biol 23: 2287-2297. [Crossref]
- Kouchi Z, Barthet G, Serban G, Georgakopoulos A, Shioi J, et al. (2009) p120 catenin recruits cadherins to gamma-secretase and inhibits production of Abeta peptide. J Biol Chem 284: 1954-1961. [Crossref]
- Carnahan RH, Rokas A, Gaucher EA, Reynolds AB (2010) The molecular evolution of the p120-catenin subfamily and its functional associations. PLoS One 5: e15747. [Crossref]
- Markham NO, Doll CA, Dohn MR, Miller RK, Yu H, et al. (2014) DIPA-family coiled-coils bind conserved isoform-specific head domain of p120-catenin family: potential roles in hydrocephalus and heterotopia. Mol Biol Cell 25: 2592-2603. [Crossref]
- Kouchi Z, Fujiwara Y, Yamaguch2021 Copyright OAT. All rights reservphatidylinositol 5-phosphate 4-kinase type II beta is required for vitamin D receptor-dependent E-cadherin expression in SW480 cells. Biochem Biophys Res Commun 408: 523-529. [Crossref]
- Kaufmann MR, Barth S, Konietzko U, Wu B, Egger S, et al. (2013) Dysregulation of hypoxia-inducible factor by presenilin/gamma-secretase loss-of-function mutations. J Neurosci 33: 1915-1926. [Crossref]
- Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M. et al. (2000) Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol 156: 965-976. [Crossref]
- Gavard J, Gutkind JS (2006) VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8: 1223-1234. [Crossref]
- Fernández-López D, Faustino J, Daneman R, Zhou L, Lee SY, et al. (2012) Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J Neurosci 32: 9588-9600. [Crossref]
- Bouleti C, Mathivet T, Coqueran B, Serfaty JM, Lesage M, et al. (2013) Protective effects of angiopoietin-like 4 on cerebrovascular and functional damages in ischaemic stroke. Eur Heart J 34: 3657-3668. [Crossref]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, et al. (2006) Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci 26: 7907-7918. [Crossref]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, et al. (2014) Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 158: 383-396. [Crossref]
- Klingener M, Chavali M, Singh J, McMillan N, Coomes A, et al. (2014) N-cadherin promotes recruitment and migration of neural progenitor cells from the SVZ neural stem cell niche into demyelinated lesions. J Neurosci 34: 9590-9606. [Crossref]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, et al. (2008) Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol 63: 520-530. [Crossref]
- Cayre M, Courtès S, Martineau F, Giordano M, Arnaud K, et al. (2013) Netrin 1 contributes to vascular remodeling in the subventricular zone and promotes progenitor emigration after demyelination. Development 140: 3107-3117. [Crossref]
- Aguirre A, Rizvi TA, Ratner N, Gallo V (2005) Overexpression of the epidermal growth factor receptor confers migratory properties to nonmigratory postnatal neural progenitors. J Neurosci 25: 11092-1110. [Crossref]
- Abe K, Takeichi M (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A 105: 13-19. [Crossref]
- Sawyer JK, Harris NJ, Slep KC, Gaul U, Peifer M (2009) The Drosophila afadin homologue Canoe regulates linkage of the actin cytoskeleton to adherens junctions during apical constriction. J Cell Biol 186: 57-73. [Crossref]
- Elia LP, Yamamoto M, Zang K, Reichardt LF (2006) p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron 51: 43-56. [Crossref]
- Yanagisawa M, Huveldt D, Kreinest P, Lohse CM, Cheville JC, et al. (2008) A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J Biol Chem 283: 18344-18354. [Crossref]
- Mori N, Kuwamura M, Tanaka N, Hirano R, Nabe M, et al. (2012) Ccdc85c encoding a protein at apical junctions of radial glia is disrupted in hemorrhagic hydrocephalus (hhy) mice. Am J Pathol 180: 314-327. [Crossref]
- Oliver C, González CA, Alvial G, Flores CA, Rodríguez EM, et al. (2013) Disruption of CDH2/N-cadherin-based adherens junctions leads to apoptosis of ependymal cells and denudation of brain ventricular walls. J Neuropathol Exp Neurol 72: 846-860. [Crossref]
- Chae TH, Kim S, Marz KE, Hanson PI, Walsh CA (2004) The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate. Nat Genet 36: 264-270. [Crossref]
- Thoreson MA, Reynolds AB (2002) Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation 70: 583-589. [Crossref]
- Wang Z, Sandiford S, Wu C, Li SS (2009) Numb regulates cell-cell adhesion and polarity in response to tyrosine kinase signalling. EMBO J 28: 2360-2373. [Crossref]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, et al. (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4: 222-231. [Crossref]
- Palacios F, Tushir JS, Fujita Y, D'Souza-Schorey C (2005) Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol 25: 389-402. [Crossref]
- Figueroa A, Kotani H, Toda Y, Mazan-Mamczarz K, Mueller EC, et al. (2009) Novel roles of hakai in cell proliferation and oncogenesis. Mol Biol Cell 20: 3533-3542. [Crossref]
- Ordóñez-Morán P, Larriba MJ, Pálmer HG, Valero RA, Barbáchano A, et al. (2008) RhoA-ROCK and p38MAPK-MSK1 mediate vitamin D effects on gene expression, phenotype, and Wnt pathway in colon cancer cells. J Cell Biol 183: 697-710. [Crossref]
- Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, et al. (2003) PHD Finger of the Chromatin-Associated Protein ING2 Functions as a Nuclear Phosphoinositide Receptor. Cell 114: 99-111. [Crossref]
- Bua DJ, Martin GM, Binda O, Gozani O (2013) Nuclear phosphatidylinositol-5-phosphate regulates ING2 stability at discrete chromatin targets in response to DNA damage. Sci Rep 3: 1-8.
- Le Jan S, Amy C, Cazes A, Monnot C, Lamande N et al. (2003) Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol 162: 1521-1528. [Crossref]
- Young JA, Ting KK, Li J, Moller T, Dunn L, et al. (2013) Regulation of vascular leak and recovery from ischemic injury by general and VE-cadherin-restricted miRNA antagonists of miR-27. Blood 122: 2911-2919. [Crossref]
- Semenza GL (2010) Vascular responses to hypoxia and ischemia. Arterioscler Thromb Vasc Biol 30: 648-652. [Crossref]
- Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, et al. (2008) Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell 3: 289-300. [Crossref]
- Young CC, van der Harg JM, Lewis NJ, Brooks KJ, Buchan AM, et al. (2013) Ependymal ciliary dysfunction and reactive astrocytosis in a reorganized subventricular zone after stroke. Cerebral Cortex 23: 647-659. [Crossref]
- Zechariah A, ElAli A, Doeppner TR, Jin F, Hasan MR, et al. (2013) Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke 44: 1690-1697. [Crossref]
- Licht T, Dor-Wollman T, Ben-Zvi A, Rothe G, and Keshet E. (2015) Vessel maturation schedule determines vulnerability to neuronal injuries of prematurity. J Clin Invest 125: 1319-1328. [Crossref]
- van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, et al. (1999) VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest 104: 1613-1620. [Crossref]
- Fernández-Lopez D, Faustino J, Derugin N, Vexler ZS (2013) Acute and Chronic Vascular Responses to Experimental Focal Arterial Stroke in the Neonatal Rat. Transl Stroke Res 4: 179-188.
- Silbereis JC, Huang EJ, Back SA, Rowitch DH (2010) Towards improved animal models of neonatal white matter injury associated with cerebral palsy. Dis Model Mech 3: 678-688. [Crossref]
- Dzietko M, Derugin N, Wendland MF, Vexler ZS, Ferriero DM (2013) Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl Stroke Res 4: 189-200. [Crossref]
- Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, et al. (2004) Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol 14: 1657-1663. [Crossref]
- Arikkath J, Peng IF, Ng YG, Israely I, Liu X, et al. (2009) Delta-catenin regulates spine and synapse morphogenesis and function in hippocampal neurons during development. J Neurosci 29: 5435-5442. [Crossref]
- Pálmer HG, González-Sancho JM, Espada J, Berciano MT, Puig I, et al. (2001) Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol 154: 369-387. [Crossref]