Abstract
Aim: The Entscheidungsproblem - the halting problem is interesting how to find stopping rule for mutagenesis, metastasis, etc. in cancer?
Materials and methods: Concerning the level of completness or incomplentness of a selected therapy is a question in cancer. It has connotations with Gὅdel’s theorem of incompletness in diseases’s definition, decission about relevant type of the therapy, etc. It is to me a question, if we are informed about all risk factors, including external factors (Feynmann’s influence functional, etc.) of the cancer in a given case ? All these problems are not only oncologic problems, but also mathematical problems in therapy.
Carcinomas are arising from epithelial tissues to higher pathological levels of malignancy through local invasion and distant metastasis. Increased expression of E-cadherin potentiates its role as an antagonist of invasion and metastasis, while reduction of its expression potentiates these phenotypes. Downregulation and mutational inactivation of E-cadherin in human carcinomas means that it is a key suppressor of the invasion and metastasis.
Results: Key orchestrators of the inflammation-mediated tumor progression, the dark side of the force, are transcription factors, cytokines, chemokines and infiltrating leukocytes. The high degree of genetic heterogeneity in tumors suggests that genetic instability is an ongoing process throughout tumor development [13]. Accumulating evidence supports the point that inflammatory mediators, some of that are direct mutagens, also directly or indirectly downregulate DNA repair pathways and cell cycle checkpoints, so destabilizing cancer cell genome and contributing to the accumulation of random genetic alterations. These in turn accelerate the somatic evolution of cancer to a genomically heterogenous population of expanding cells naturally selected for their ability to proliferate, invade and evade host defenses. This is a dual function of inflammatory cells and mediators. The challenge now is to identify the mechanisms triggering a ‘bright’ inflammatory response leading to tumor inhibition, while neutralizing the protumor actions of the dark side. In anti-cancer therapy of post-genomic era may be fruitful the network-based modeling approach, in general mathematical methods too. To give the process a kick-start, the vaccine is being combined with low doses of chemotherapy, in order to kill some of the tumor cells and disinhibit the immune system. The investigators behind the trial believe the vaccine could prove effective for all types of solid tumors, and are testing its safety and efficacy on patients whose cancer has been diagnosed as terminal. Cells do not make mutations because they have only to repair DNA. Rather, this mechanism regulate production of mutations, making more during stress, when cells are poorly adapted to their coding environments, most likely to benefit from mutations.
Conclusion: There is a big therapeutical potential in using mathematical analysis to map the correlations leading to carcinogenesis and its therapy.
Key words
limit, stress, gene network, maximal element, program, suppressors, mutation, adaptation
Introduction
Tumors are organs with nonlinear mathematics. The recruited normal cells form tumor-associated stroma as active participants of tumorigenesis. Tumors are more than insular masses of proliferating cancer cells. The most fundamental trait of cancer cells involves their ability to sustain chronic proliferation. The gene networks functions in causing mutations during repair of double-stranded breaks in the DNA of stressed cells. A network of 93 genes is funnelling into 3 nodes that go down the mutagenesis pathway.
Similar to how vaccines protect us from certain infections, the treatment attempts to recruit the body’s own immune system to attack and destroy specific entities – in this case cancer cells. Normally, the various components of the immune system – which include white blood cells such as T cells – protect against cancer by killing tumor cells, although some tumors are able to evade these natural defenses.
When this occurs and cancer develops to an advanced stage, the immune system is often supressed. Numerous factors are thought to be responsible for this effect, ranging from tumor cells’ ability to damage immune cells, to a decrease in white blood cell production when cancer spreads to the bone marrow.
Vaccines typically work by injecting a patient with small amounts of antigens, substances that are capable of eliciting an immune response, which stimulate the body to produce antibodies that specialize in the labeling and destruction of that particular entity. The ability to generate these antibodies is retained for a period of time, meaning that the immune system can then fend off future cases of the same illnesses.
Materials and methods
Cancer cells achieving the capabability to sustain proliferative signaling due producing growth factor ligands themselves, via the expression of cognate receptors, resulting in autocrine proliferative stimulation. Cancer cells send signals to stimulate normal cells within the supporting tumor-associated stroma, reciprocate by supplying the cancer cells with growth factors. Receptor signalling is deregulated by elevating the levels of receptor proteins manifested at the cancer cell surface. Such cells are hyper responsive by stress to otherwise-limiting amounts of growth factor ligand. High-throughput DNA sequencing analyses of cancer cell genomes showed somatic mutations in human tumors predicting constitutive activation of signalling circuits triggered by activated growth factor receptors. The advantages to tumor cells of activating upstream (receptor) versus downstream (transducer) signalling have functional impact of crosstalk between multiple pathways radiating from growth factor receptors. In activating is important the effect of ambiguity on the perceived probability is often modeled as the SV by using a power function: SV =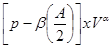 , where
, where 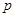 is the objective probability,
is the objective probability, 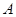 is the ambiguity (the fraction of the total probability is unknown),
is the ambiguity (the fraction of the total probability is unknown), 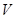 is the amount which can be won,
is the amount which can be won, 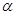 and
and 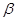 are subject-specific risk and ambiguity parameters. The effect of ambiguity on the subjective probability is exponential rather than linear SV=
are subject-specific risk and ambiguity parameters. The effect of ambiguity on the subjective probability is exponential rather than linear SV=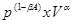 , where
, where 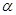 is a risk preference parameter,
is a risk preference parameter, 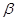 is ambiguity aversion parameter [1].
is ambiguity aversion parameter [1].
Tumors - organs with nonlinear mathematics
Also paradoxical responses reveal intrinsic cellular defence mechanisms eliminating cells under excessive levels of signalling. The relative intensity of oncogenic signalling in cancer represent compromises between maximal mitogenic stimulation and avoidance of antiproliferative defences. Cancer cells implement powerful programs that negatively regulate cell proliferation. These programs depend on the activity of tumor supressor genes operating in various ways to limit cell growth and proliferation. The two prototypical tumor suppressors encode the RB (retinoblastoma-associated) and TP53 proteins operating as central nodes within two key complementary cellular regulatory circuits that govern the decisions of cells to proliferate or, activate senescence and apoptotic programs. RB transduces growth-inhibitory signals originating outside of the cell, TP53 receives inputs from stress and abnormality sensors within the cell’s intracellular operating systems. These two canonical supressors of proliferation: RB and TP53 have preeminent importance in regulating cell proliferation, each operates as part of larger gene network of stress, mutation and adaptation responses wired for functional redundancy [2]. The regulation of proliferation we model with the q-distribution may be the exact posterior over 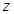 or an approximation. The free energy
or an approximation. The free energy 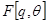 is related to Kullback-Liebler divergence between the q-distribution
is related to Kullback-Liebler divergence between the q-distribution 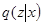 and the true posterior
and the true posterior 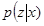 as follows:
as follows: 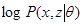 =
= 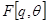 +
+ 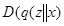
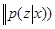 . The Kullback-Liebler divergence is a measure of the difference between probability distributions. If this zero of the distributions are equal, and positive otherwise, thus the free energy
. The Kullback-Liebler divergence is a measure of the difference between probability distributions. If this zero of the distributions are equal, and positive otherwise, thus the free energy 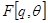 is a lower bound on the complete data log likelihood. The EM algorithm can be expressed as:
is a lower bound on the complete data log likelihood. The EM algorithm can be expressed as:
E-Step: 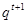
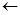 arg
arg 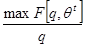
M-Step: 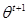
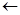 arg
arg 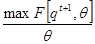
Both the E-Step and the M-Step maximize the same objective function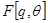 . The local maximum of the free energy will only correspond to a local maximum of the expected complete log likelihood when
. The local maximum of the free energy will only correspond to a local maximum of the expected complete log likelihood when 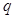 is a true maximizer of
is a true maximizer of 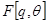 .
.
The contact inhibition is an in vitro a mechanism operating in vivo to ensure normal tissue homeostasis, which is abroagated during the tumorigenesis. The mechanistic basis for this mode of growth control involving two elements. First mechanism involves Merlin, the cytoplasmic NF2 gene product, orchestrates contact inhibition via coupling E-cadherin (cell-surface adhesion molecules) to the EGF receptor (transmembrane receptor tyrosine kinases). Merlin strengthens the adhesivity of cadherin-mediated cell-to-cell attachments. By sequestering growth factor receptors limits their emitting of mitogenic signals. The second mechanism of contact inhibition involves the LKB1 epithelial polarity protein, helps maintain tissue integrity. If the mitogenic effects are upregulated, LKB1 overrule the powerful Myc oncogene. When LKB1 expression is suppressed, epithelial integrity is destabilized, epithelial cells are susceptible to Myc-induced transformation. LKB1 also function as a suppressor gene of inappropriate proliferation. TGF-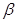 is known also for its antiproliferative effects. But in late-stage tumors, TGF-
is known also for its antiproliferative effects. But in late-stage tumors, TGF-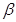 signaling is redirected away from suppressing cell proliferation and instead activates a cellular program, the epithelial-to-mesenchymal transition (EMT) which confers to cancer traits with high-grade malignancy [3].
signaling is redirected away from suppressing cell proliferation and instead activates a cellular program, the epithelial-to-mesenchymal transition (EMT) which confers to cancer traits with high-grade malignancy [3].
Tumors strategies to limit apoptosis
Tumor cells produce many strategies to limit apoptosis. The plurality of apoptosis-avoiding mechanisms probably is given by the diversity of apoptosis-inducing signals during evolution of cancer cells to the malignant state. Similarly to apoptosis, the autophagy machinery has both regulatory and effector components. The signaling pathway involving the PI3-kinase, AKT, and mTOR kinases are stimulated by survival signals to block apoptosis, also inhibits autophagy. Stress-transducing BH3-proteins (Bid, Bad, Puma, etc.) can induce apoptosis or autophagy depending on the physiologic state of the cell. Induction of autophagy can serve as a barrier to tumorigenesis which may operate independently or in concert with apoptosis. Nutrient starvation, radiotherapy, and cytotoxic drugs may induce elevated levels of autophagy that are cytoprotective for cancer cells. Stressed cancer cells are going to shrink via autophagy to a state of reversible dormancy. Like the TGF-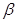 signaling, (tumor suppressing in early stages of tumorigenesis, and tumor-promoting in later), autophagy induces inhibitional antagonism in tumor cells and tumor progression. In apoptosis, a dying cell contracts into a almost-invisible corpse, which is consumed by neighbors (cannibalism). The necrotic cells explode and releasing their contents into the local tissue microenvironment. Cell death by necrosis is under genetic control, rather than being a random. Necrotic cell death releases proinflammatory signals into tissue microenvironment, while apoptosis and autophagy do not. Necrotic cells can recruit inflammatory cells of the immune system and can be actively tumor promoting.
signaling, (tumor suppressing in early stages of tumorigenesis, and tumor-promoting in later), autophagy induces inhibitional antagonism in tumor cells and tumor progression. In apoptosis, a dying cell contracts into a almost-invisible corpse, which is consumed by neighbors (cannibalism). The necrotic cells explode and releasing their contents into the local tissue microenvironment. Cell death by necrosis is under genetic control, rather than being a random. Necrotic cell death releases proinflammatory signals into tissue microenvironment, while apoptosis and autophagy do not. Necrotic cells can recruit inflammatory cells of the immune system and can be actively tumor promoting.
In our model of inhibitional antagonism the values of 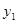 and
and 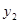 are transformed through a nonlinear activation function
are transformed through a nonlinear activation function 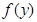 before they inhibit each other:
before they inhibit each other:
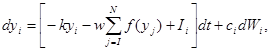 integration starts from
integration starts from  , an input unit with mean activity
, an input unit with mean activity  , and independent white noise fluctuations
, and independent white noise fluctuations 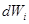 of amplitude
of amplitude 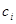 . The next part of the inhibitory antagonism’s cascade, when these units also inhibit each other with a connection weight
. The next part of the inhibitory antagonism’s cascade, when these units also inhibit each other with a connection weight 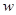 ,
, 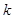 denotes the decay (disintegration, inflation) rate of the accumulated activity with leak (entropy, deflation),
denotes the decay (disintegration, inflation) rate of the accumulated activity with leak (entropy, deflation), 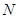 means the number of alternatives.
means the number of alternatives.
Performance and dynamics of choice models suggests that in some cases the balance of inhibition and decay is not optimizing the performance, but rather it may be more profitable increase the inhibition parameter 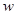 , which increase inhibition of accumulators
, which increase inhibition of accumulators 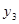 ,
, 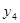 ,
, 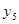 and thus prevent from the competition with
and thus prevent from the competition with 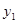 and
and 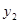 (for
(for 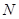 = 5 alternatives). This type of preemptive discrimination in favor of ‘pointer states’, which is suppressing further competitive behavior, is known from the Quantum Darwinism as a mechanism of “inquisition” [4].
= 5 alternatives). This type of preemptive discrimination in favor of ‘pointer states’, which is suppressing further competitive behavior, is known from the Quantum Darwinism as a mechanism of “inquisition” [4].
Another barrier to cancer pathogenesis represents the intrinsic apoptotic program. Culminates in activation of normally latent protease (caspases 8 and 9) which initiate a cascade of proteolysis involving effector caspases for the execution phase of apoptosis (cell is disassembled and consumed) both by its neighbors and by special phagocytic cells. The cellular conditions that trigger apoptosis, involves abnormality sensors paying key role in tumor development. Most known is a DNA-damage sensor that functions via the TP53 tumor suppressor. During this process TP53 induces apoptosis by upregulating expression of the Noxa and Puma BH3-only proteins, in response to DNA breaks and other chromosomal abnormalities.
Cancer as universal turing machine
Cancer cells require unlimited replicative potential of the Tumor as Universal Turing Machine (UTM) to generate macroscopic tumors. It is in contrast to the behavior of the normal cells, which are able to pass through only a limited number of successive cell growth-and-division cycles given by the Hayflick limit. This limitation has two prominent barriers to proliferation: senescense (an irreversible entrance into a nonproliferative but viable state) and crisis (involves cell death). In some cases cells emerge from a crisis population and exhibit unlimited replicative potential termed immortalization. The telomeres (protecting the ends of chromosomes) are centrally involved in unlimited proliferation. If telomeres shorten, or losing the ability to protect the ends of chromosomal DNAs from the end-to-end fusions, they generate unstable dicentric chromosomes that threatens cell viability. The length of telomeric DNA in a cell dictates how many successive cell generations can pass through before telomeres are eroded and have lost their protective functions, triggering entrance into crisis. The two barriers to proliferation: senescence and crisis/apoptosis are crucial anticancer defenses and are hard-wired into our cells to impede the outgrowth of clones of preneoplastic and neoplastic cells. When neoplasias exhaust their basis of replicative doublings, then are stopped by one of these barriers. The telomere shortening can be seen as a clocking device determining the limited replicative potential of normal cells given by Hayflick limit which must be overcome by cancer cells.
The cell senescence is a protective barrier to neoplastic expansion triggered by proliferation-associated abnormalities, high levels of oncogenic signaling, subcritical shortening of telomeres The multistep tumor progression is due to inability to express significant levels of telomerase. Some human neoplasias may be aborted by telomere-induced crisis before macroscopic neoplastic growths. Tumor progression we can represent as start with a topological space  and consider the sheaf of all continuos real-valued function defined on
and consider the sheaf of all continuos real-valued function defined on 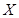 . This assoaciates to every open set
. This assoaciates to every open set 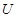 in
in 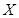 the set
the set 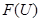 of real-valued continuous functions defined on
of real-valued continuous functions defined on 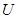 . Whenever
. Whenever 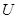 is a subset of
is a subset of 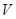 , we have a restriction map from
, we have a restriction map from 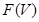 to
to 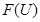 . If we interpret the topological space
. If we interpret the topological space 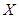 as a category, with open sets being the objects and a polymorphism from
as a category, with open sets being the objects and a polymorphism from 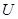 to
to 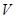 if and only if
if and only if 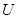 is a subset of
is a subset of 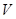 , then
, then 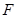 is revealed as a contravariant functor from this category into the category of sets. In general, every contravariant functor from category
is revealed as a contravariant functor from this category into the category of sets. In general, every contravariant functor from category 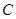 to the category of sets is therefore called a pre-sheaf of sets on
to the category of sets is therefore called a pre-sheaf of sets on 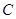 . Functor
. Functor 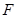 has a special property: if you have an open covering
has a special property: if you have an open covering 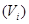 of the set
of the set 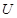 , and you are given mutually compatible elements of
, and you are given mutually compatible elements of 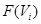 , then there exists precisely one element
, then there exists precisely one element 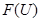 which restricts to all the given ones. This is the defining property of a sheaf and a Grothendick topology on
which restricts to all the given ones. This is the defining property of a sheaf and a Grothendick topology on 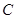 is an attempt to capture the essence of what is needed to define sheaves on
is an attempt to capture the essence of what is needed to define sheaves on 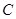 .
.
The absence of TP53-mediated surveillance of genomic integrity permits other neoplasias to survive initial telomere erosion and chromosomal breakage-fusion-bridge (BFB) cycles. The genomic alterations caused by these BFB cycles, using deletions, increase the mutability of genome. Impaired telomere function foster tumor progression lacks both p53 and telomerase function and these defects enhance human tumorigenesis. The delayed acquisition of telomerase function generates tumor-promoting mutations, and confers the unlimited replicative capacity generating tumors. Telomerase exerts functions relevant to cell proliferation, but unrelated to telomere maintenance. This noncanonical role of telomerase is particularly given by its protein subunit TERT.
Carcinomas are arising from epithelial tissues to higher pathological levels of malignancy through local invasion and distant metastasis. Increased expression of E-cadherin potentiates its role as an antagonist of invasion and metastasis, while reduction of its expression potentiates these phenotypes. Downregulation and mutational inactivation of E-cadherin in human carcinomas means that it is a key suppressor of the invasion and metastasis. The process of invasion and metastasis is schematized as a sequence of discrete steps termed the invasion-metastasis cascade leading to colonization. A developmental regulatory program means the epithelial-mesenchymal transition (EMT) by which transformed epithelial cells acquire capability to invade and disseminate. Some transcription factors directly repress E-cadherin gene expression, what is depriving neoplastic epithelial cells of key suppressor of motility and invasiveness. Crosstalk between cancer cells and cells of neoplastic stroma are involved in the acquired capability for invasive growth and metastasis [3].
Modeling cancer metastases
For modeling cancer metastases spread there is several new methodologies. The nodes of a cancer metastasis network represents the distant sites where metastases could arise for a given tumor type. The size of each node represents its conditional incidence or hazard. The incidence hazard function is 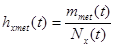 where
where 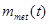 is the number of diagnoses of metastasis met at time
is the number of diagnoses of metastasis met at time 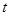 and
and 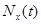 is the number of patients remaining at time
is the number of patients remaining at time 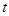 for primary tumor type
for primary tumor type 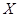 . (Chen et al., 2009) The cummulative hazard from
. (Chen et al., 2009) The cummulative hazard from 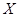 and met pair is
and met pair is
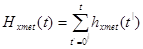 to quantify the dynamics of metastasis development, we need the incidence of metastases in terms of co-occurence at every point of time. This allows to istablish links between the primary tumor and metastasis sites, as between different metastasis sites for multiple cases.
to quantify the dynamics of metastasis development, we need the incidence of metastases in terms of co-occurence at every point of time. This allows to istablish links between the primary tumor and metastasis sites, as between different metastasis sites for multiple cases.
With the fractional method as a baseline for comparison was developed an algorithm for predicting future sites of metastases using cancer metastasis networks. These networks are entities on which the metastatic disease of individual patients evolve, and are able to incorporate temporal dynamics, and subtle relational properties [5-9]. Between anti-inflammatory receptors on macrophages of particular interest is CD200, whose by the endogenous ligand CD200L sends stop signal to macrophages suppressing production of proinflammatory mediators. CD200L expressed on activated immune cells provides a mechanism to dampen macrophage activation after initiation of the inflammatory response [10]. Another receptor involved in negative regulation of inflammation is the recepteur d’origine nantis (RON), or stem cell-derived tyrosine kinase (STK) receptor in the mouse. The ligand for this receptor is macrophage-stimulating protein (MSP), a serum protein generated during the coagulation cascade. Even TLRs have the capacity of inducing expression of anti-inflammatory mediators, like IL-10. The signaling pathway used by TLRs to activate expression of pro- and anti-inflammatory cytokines diverges at the level of the TRAF3 and TRAF6 proteins, because TRAF3 is critical for induction of IL-10 expression. In its absence, expression of the TRAF6-dependent proinflammatory cytokines IL-6 and IL-12 is dramatically upregulated.
Recent experiments have shown that nutrient starvation may induce intense autophagy which is causing cancer cells to shrink and adopt a state of reversible dormancy. These cells may exit from this state and resume active growth and proliferation after changes in tissue microenvironment (access to more nutrients). Each type of disseminated cancer cell needs to develop its own set of ad hoc solutions to the problem of thriving in the microenvironment of foreign tissue [3].
The regulatory programs of metastatic colonization
Macroscopic metastases may erupt decades after a primary tumor was surgically removed, or pharmacologically destroyed. These metastatic growths evidently reflect dormant micrometastases that have solved, after much trial and error, the complex problem of tissue colonization. This may support our hypothesis that tumor like an organ can be seen as a UTM programmed to make decisions outgoing from the Hayflick limit and trying to solve the Entscheidungsproblem. These adaptations require hundreds of distinct colonization programs, each dictated by the type of disseminating cancer cells and the nature of the tissue microenvironment. The colonization we can model with vectors. Let 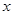 be a vector of observed variables,
be a vector of observed variables, 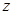 be a vector of latent variables, and
be a vector of latent variables, and 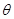 be the model parameters. Lwt
be the model parameters. Lwt 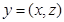 be a vector of all variables in the model. If
be a vector of all variables in the model. If 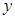 were completly observed we could apply standard maximum likelyhood estimation to obtain:
were completly observed we could apply standard maximum likelyhood estimation to obtain:
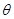 = argmax
= argmax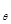 log
log 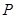 (
(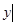
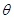 ). The
). The 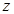 is unobserved,
is unobserved, 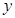 becomes a random variable and there must be apply the Expectation Maximization (EM) algorithm. For more complex models, the free energy approach leads to more flexible model fitting. The standard EM is equivalent to performing coordinate ascent on the energy function
becomes a random variable and there must be apply the Expectation Maximization (EM) algorithm. For more complex models, the free energy approach leads to more flexible model fitting. The standard EM is equivalent to performing coordinate ascent on the energy function
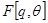 =
= 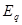
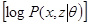 +
+ 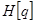 , where
, where 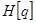 =
= 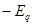
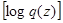 .
.
Beyond the timing we must know where cancer cells achieve the ability to colonize foreign tissue as macroscopic tumors. It may be during primary tumor formation when these cells enter into the circulation and are fortuitously endowed with capability to colonize distant tissues. The ability to colonize may also develop in response to the selective pressure on disseminated cancer cells to adapt to growth in foreign tissue microenvironment [2]. Tissue–specific colonization programs are evident in cells within primary tumor, may originate from emigrants returned home.
The phenotypes and underlying gene expression programs of cancer cells within primary tumors may be modified by reverse migration of their distant metastatic progeny. In this self-seeding process the supportive stroma (arising in a primary tumor) contributing to acquisition of its malignant traits make possible reseeding and colonization by circulating cancer cells from metastatic lesions. The regulatory programs enabling metastatic colonization emerging in an important agenda for future research. Above mentioned multitude of colonization programs are unlikely to depend only on cell-autonomous processes. The process of colonization probably encompass a large number of cell-biological programs, which are nonlinear and diverse. The numerous signaling molecules affecting cancer cells as nodes and branches of the elaborate integrated circuits which are reprogrammed derivatives of the circuits operating in normal cells.
Bacterial starvation and general stress responses also promote mutagenesis during stress. Include the stringent and competence starvation-stress responses in Bacillus subtilis, and RpoE membrane-protein stress response in E.coli. These examples illustrate the multiple evolutions of mechanisms that couple genomic instability pathways with stress responses and stress. The importance of all of these is that genetic diversity is generated preferentially when cells are maladapted to their environment: when stressed.
Mechanisms promoting mutation in stress
Mechanisms that promote mutation during stress by coupling mutagenesis to stress responses can enhance the ability of cells to evolve rapidly, responsively to environment, specifically when they are maladapted. This responsive gene-diversification strategy is similar to how protein diversity is maximal during stress when chaperons are less available, potentially promoting evolvability. Generation of phenotypic and genetic variation may be means by which organisms accelerate evolution responsively to their environments. Both stress-induced mutagenesis and phenotypic diversification contribute to observations of climate/heat stress-induced enhanced expression of genetic variation, leading to rapid evolution.
Interestingly, the halting problem still applies to machines, although they can determine whether machines with equivalent halting oracles will themselves halt. Since a conventional Turing machine cannot solve the halting problem [11]. But any coupled machine with a finite input stream can be simulated by the UTM since the data can be written on the machine’s tape before it begins operation. From dynamic systems we often decide almost an arbitrarily way, when a system will be closed in order to handle it. The chain of such external systems, potentially infinite (by loops) can create a non-linear system which could truly be more complex and more powerful. Because we are interested in the set of Turing degrees of the weights of a gene network, it is necessarily to use the notion of maximal element. Let be a partially ordered set (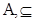 ). Then an element
). Then an element 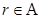 is said to be maximal if, for all
is said to be maximal if, for all 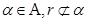 . A real number
. A real number 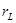 is computable if and only if it is the limit of an effectively converging computable sequence of rational numbers. The Turing degree of a real number
is computable if and only if it is the limit of an effectively converging computable sequence of rational numbers. The Turing degree of a real number 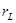 is defined as the Turing degree of its binary expansion. If
is defined as the Turing degree of its binary expansion. If 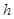 is used as a weight, where
is used as a weight, where 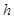 is the Chaitin constant
is the Chaitin constant  defined as the probability of halting of a Turing machine tumor
defined as the probability of halting of a Turing machine tumor 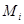 for an input
for an input 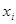 , gene network
, gene network 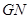 is going to compute definable languages but non-Turing computable. It is evident that
is going to compute definable languages but non-Turing computable. It is evident that 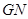 computes
computes 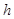 and those in its same complexity degree. If the set of maximals of
and those in its same complexity degree. If the set of maximals of 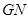 has only
has only 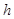 , that is
, that is 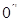 .
.
Accordingly, the new vaccine currently being tested is comprised of small fragments of an enzyme found in cancer cells. Called human telomerase reverse transcriptase (hTERT), this regulates the length of the protective caps on chromosomes called telomeres, enabling the cells to divide continuously. Scientists are hopeful that this will stimulate patients’ immune systems to produce antibodies that can target this enzyme, thereby facilitating the destruction of cancer cells [12].
Conclusion
Evidence began to accumulate in the ends of the 1990s that the infiltration of neoplastic tissues by cells of the immune system serves, counter intuitively, to promote tumor progression. Infiltrating cells of the immune system are increasingly accepted to be generic constituents of tumors. In most of neoplastic lesions can be found inflammatory cells (leucocytes) operating in conflicting ways: both tumor-antagonizing and also tumor-promoting. These capabilities are showing the dichotomous roles of an immune system which both antagonizes and enhances tumor development and progression. To quantify the co-occurrence of metastases there can be used a measure of the 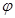 -correlation (Pearson’s correlation between dichotomous variables) as:
-correlation (Pearson’s correlation between dichotomous variables) as:
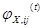 =
=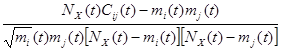
where 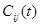 is the number of co-occurrence at time
is the number of co-occurrence at time 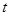 . Then
. Then 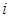 and
and 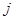 are particular sites of metastasis (or the primary tumor itself), and
are particular sites of metastasis (or the primary tumor itself), and 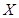 represents the primary tumor type. The
represents the primary tumor type. The 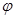 -correlation tends to be better measure for analysing links across multiple cancer metastasis networks [8].
-correlation tends to be better measure for analysing links across multiple cancer metastasis networks [8].
Alan Turing was perfectly aware that generating the capacity to does not necessarily generate the capacity to feel. The TT-passing model may not to be purely computational, it may be both computational and dynamic, but it is still only generating and explaining our doing capacity. It may or may not feel. Explaining how and why we can do what we can do has to come to be called the “easy” problem of cognitive science. The “hard” problem is explaining how and why we feel. The current state of the art in the attempt to give a causal explanation of how and why we feel rather than just do is solved thanks to groundbreaking work of Susan Rosenberg [2].
The puzzling response of bacteria to stress and the mutations that result she solved by describing most of the numbers of an elaborate gene network that functions in causing mutations during repair of double-stranded breaks in the DNA of stressed cells. We know the 93 genes more than half of which are funnelling into three nodes that go down the mutagenesis pathway. It has shown that the rate of mutation can be increased in response to stress such as starvation or environmental challenges. This mechanism appears to regulate production of mutations, making more during stress, when cells are poorly adapted to their environments, and most likely to benefit from mutations. Large fractions of the network work upstream of the activation of the stress response, showing that these proteins sense the stress. They identified specific pathways through which the proteins sense the environment and connected them to the molecular mechanism that promotes the mutations. The findings reveal key factors about the cells, like stress-response regulators, which acts as key network hubs. Most of the proteins in the network deal with whether or not the cells feel the stress. It’s a confirmation of the regulation of mutagenesis by stress responses, which causes mutations specifically when cells are maladapted to their environment when mutations might allow to the cell to adapt.
2021 Copyright OAT. All rights reserv
Cancer cells constitute one of main exception to the limits on cell division. Probably, the Hayflick limit exists principally to help prevent cancer. Once the cell reaches this limiting Hayflick number of divisions, the former tumor will no longer be able to reproduce and the cells will die off. Cancers become problems after having reactivated telomerase-encoding genes. Cells that have found a way around the limit are termed immortal. They may still die, the group of immortalized cells from cell division of an immortal cell has no limit as to how many times cell division might take place among the cells that constitute a such group of immortalized cells. Probably some or all cancers start off as stem cells that become genetically damaged. This would mean they already aren’t limited by the Hayflick limit and can easily metastasize into the pool of cells in their final cell type destination.
In fact, replicative senescence (RS) is also likely an anti-cancer mechanism. One hypothesis is that while RS evolved as an anti-cancer mechanism, the accumulation of senescent cells contributes to aging. It is possible that stress and insults trigger cell senescence in vivo. Because of telomere shortening is the main cause of RS in human fibroblasts.
Key orchestrators of the inflammation-mediated tumor progression, the dark side of the force, are transcription factors, cytokines, chemokines and infiltrating leukocytes. The high degree of genetic heterogeneity in tumors suggests that genetic instability is an ongoing process throughout tumor development [13].
Accumulating evidence supports the point that inflammatory mediators, some of that are direct mutagens, also directly or indirectly downregulate DNA repair pathways and cell cycle checkpoints, so destabilizing cancer cell genome and contributing to the accumulation of random genetic alterations. These in turn accelerate the somatic evolution of cancer to a genomically heterogenous population of expanding cells naturally selected for their ability to proliferate, invade and evade host defenses. This is a dual function of inflammatory cells and mediators. The challenge now is to identify the mechanisms triggering a ‘bright’ inflammatory response leading to tumor inhibition, while neutralizing the protumor actions of the dark side. In anti-cancer therapy of post-genomic era may be fruitful the network-based modeling approach, in general mathematical methods too. To give the process a kick-start, the vaccine is being combined with low doses of chemotherapy, in order to kill some of the tumor cells and disinhibit the immune system. The investigators behind the trial believe the vaccine could prove effective for all types of solid tumors, and are testing its safety and efficacy on patients whose cancer has been diagnosed as terminal.
Cells do not make mutations because they have only to repair DNA. Rather, this mechanism regulate production of mutations, making more during stress, when cells are poorly adapted to their coding environments, most likely to benefit from mutations. We are outgoing from the network-based modelling approach. A newly developed vaccine that may prove effective at destroying tumors in cases where all other treatments have failed is currently being trailed in the UK at Guy’s Hospital in London.
We are trying outgoing from own observations and new ideas concerning the relationship between the immunological traits and mathematical aspects of tumorigenesis. It is clear, that mathematical methods are allowing for new definitions of above correlations instead the big amounts of experiments on animals, which often are only proving again already known references cited by experimentators. There is a big therapeutical potential in using mathematical analysis to map the correlations leading to carcinogenesis and its therapy.
Acknowledgements
The author gratefully acknowledges the assistance of Dr. Marta Ballova, Ing. Konrad Balla, Livuska Ballova, and Ing. Jozef Balla.
References
- Levy I, Snell J, Nelson AJ, Rustichini A, Glimcher PW (2010) Neural representation of subjective value under risk and ambiguity. J Neurophysiol 103: 1036-1047. [Crossref]
- Al Mamun AAM, Lombardo M-J, Shee C, Lisewski AM, Gonzalez C, et al, (2012) Identity and Function of a Large Gene Network Underlying Mutagenic Repair of DNA Breaks. Science 338: 1344-1348. [Crossref]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674. [Crossref]
- Zurek WH (2009) Quantum Darwinism. arXiv: 0903.5082v 1 [quant-ph].
- Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, et al. (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440: 692-696. [Crossref]
- Loscalzo J, Kohane J, Barabasi AL (2007) Human disease classification in the postgenomic era: a complex system approach to human pathobiology. Mol Syst Biol 3: 124. [Crossref]
- Lee DS, Park J, Kay KA, Christakis NA, Oltvai ZN, et al. (2008) The implications of human metabolic network topology for disease comorbidity. Proc Natl Acad Sci USA 105: 9880-9885. [Crossref]
- Chen LL, Blum N, Christakis NA, Barabasi AL, Deisboeck TS (2009) Cancer metastasis networks and the prediction of progression patterns. British J Cancer 101: 749-758.
- Huang Q, Li F, Liu X, Li W, Shi W, et al. (2011) Caspase-3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 77: 860-866. [Crossref]
- Karin M, Lawrence T, Nizet V (2006) Innate Immunity Gone Awry: Linking Microbial Infections to Chronic Inflammation and Cancer. Cell 124: 823-835. [Crossref]
- Turing AM (1936) On computable numbers, with an application to the Entscheidungsproblem. Proc London Math Soc 42: 230-265.
- Taub B (2016) Human trials for a radical new cancer vaccine just began. IFL Science.
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30: 1073-1081. [Crossref]