Over the last decade, management of melanoma has dramatically evolved from chemotherapy through targeted molecular therapy (BRAF V600E signaling) and, currently, immunotherapy (checkpoint inhibitors, immunogenic oncolytic viruses). Response, time to progression and survival has improved for many melanoma patients undergoing targeted therapy, but insensitive population subsets, adaptive resistance and toxic side effects limit therapeutic benefit. Previous studies have shown a correlation between VigilÒ engineered autologous tumor cell (EATC) immunotherapy induced circulating activated T-cells responsive against autologous tumor cells and survival prolongation. We now assess the safety and response to Vigil (1 x 107 cells/ intradermal injection monthly x 4-12 months) in 12 patients with advanced metastatic melanoma in comparison with 12 who underwent similar standard of care autologous tumor harvest but received other treatment regimens rather than Vigil. None of the patients experienced ≥ Grade 3 treatment-related toxicity. Two Grade 2 adverse events (AE) (fatigue, irritability) and local regional Grade 1 AE (injection site erythema, induration, rash, skin hypopigmentation) in 19 of 63 injections were observed. IFN-γ ELISPOT analysis (PBMC) showed the induction of T-cell activation from 0-1 at baseline to 78 spots/106 cells post first cycle of Vigil. Median survival of Vigil treated patients was 20 months compared to 7 months (Kaplan Meier analysis, log rank p=0.00009). In conclusion, preliminary evidence of safety and activity of Vigil supports further clinical evaluation in advanced melanoma.
Melanoma is the prototype of “immunogenic tumors” with a heightened expression of cancer-testes antigens (e.g., MAGE-A3. MAGE-A1, NY-ESO-1 and SSX-2), a high tumor mutation burden (TMB) leading to an increased number of tumor-specific epitopes, and clinically a reproducible response rate to immunotherapies [1-4] particularly to the recently FDA approved immune checkpoint inhibitors. One of these inhibitors is ipilimumab (Yervoy; a human monoclonal antibody (hMAb) CTLA-4 inhibitor), which was FDA approved in 2011 for patients with advanced, unresectable Stage III and IV melanoma [5]. Results show improvement in recurrence-free survival (RFS) as compared to placebo in the EORTC trial 18071 (HR 0.75, 95 % CI 0.64 – 0.90), [6]. Subsequently, pembrolizumab (Keytruda), a hMAb PD-1 inhibitor, elicited response rates of 36% [7] and has since proven to be superior to chemotherapy and single agent ipilimumab in patients with advanced melanoma [8-10] as has nivolumab (Opdivo) [11]. However, >60% of melanoma patients do not achieve a significant response to a single agent checkpoint inhibitor and subsets of patients (i.e. PD-L1-; low TMB) respond less favorably. The combination of mechanistically different immune checkpoint inhibitors does elicit higher response rates. For example, in a randomized comparision of nivolumab alone to ipilimumab alone or to the combination in treatment-naïve patients with unresectable stage III or IV melanoma, the combination achieved an ORR of 57.6% (compared to 43.7% with nivolumab and 19% with ipilimumab) with a durable response of 11.5 months, but also resulted in 55% treatment-related ≥Grade 3 toxicities leading to discontinuation in 36.4% of patients [9]. Although these data confirm the effectiveness of immunotherapy in advanced melanoma, they also highlight the need for further development of novel and/or combinatory immunotherapies with increased, predictable effectiveness at a lower risk of toxicity. Talimogene laherparepvec (T-VEC), an intra-tumorally administered genetically-modified, immune-enhanced H. simplex type I virus, is effective in advanced melanoma [12] but the FDA indication is limited to patients with unresectable stages IIIb, IIIc or IVM1a disease based on stage-related efficacy shown in Phase III testing [13,14].
Vigil is a DNA engineered autologous tumor cell (EATC) immunotherapy. It contains a dual vector; a bi-shRNA targeting furin the pro-protein convertase that activates the immunosuppressive TGF-beta 1 and 2 and the gene encoding hGM-CSF. A phase I clinical trial of Vigil in patients with heavily pretreated advanced solid tumors showed a significant survival benefit which correlated with induction of an immune response measured by the interferon gamma (IFN-γ) ELISPOT assay. We now update the results of Vigil clinical activity in patients with advanced melanoma.
The method and mechanism of construction and manufacturing of Vigil (formerly known as FANG) has previously been described [15,16]. The Vigil vector encodes for GM-CSF expressive cDNA and the bi-shRNAfurin in autologous tumor cells. Following protocol-specific informed consent, tumor tissue is harvested, placed in sterile media and delivered to the Gradalis, Inc. manufacturing facility (Carrollton, TX, USA). Vigil is manufactured over 2 conservative days. (Since these studies were completed, the manufacturing has been modified and, following FDA discussion, now utilizes Gentamicin in the sterile media in order to reduce contamination risk. First, autologous tumor cells are dissociated into a single-cell suspension, followed by electroporation (which allows cell transfection with the plasmid), and overnight incubation. Then the cells are irradiated, placed into the final vials, cryopreserved, and undergo release testing. Following product release in accordance with Quality Assurance guidelines, patients are registered for treatment every 4 weeks with 1.0 x 107 cells/injection of Vigil.
Study design
This follow-up includes all Vigil treated melanoma patients enrolled in both the Phase I solid tumor trial [15] and a Phase II trial of Vigil in patients with advanced or recurrent melanoma. The primary objective of the Phase I trial was to determine safety following the administration of Vigil (EATC). The primary objective of the non-randomized Phase II open label trial was to evaluate the effect of Vigil on immune stimulation in patients with melanoma and to assess survival efficacy in comparison with historical data.
Secondary objectives were to expand the Phase I safety evaluation of Vigil immunotherapy in patients with advanced solid tumors without alternative standard therapy options and to evaluate effectiveness based on IFN-γ ELISPOT induction/conversion and on survival in both the Phase I melanoma and Phase II patients.
Depending on the manufactured cell yield from the harvested tumor so as to produce a minimum dose criterion of 1 x 107 cells/ml (and 2.5 x 107 cells/ml in Phase I), patients were eligible to receive a maximum of 12 intradermal injections. Each injection was administered monthly, alternating between the right and left upper arms. Safety assessment included physical examination, performance status, weight, height, temperature, blood pressure and pulse, as well as toxicity profile. Laboratory assessment, blood immune biomarker assessment, response rate [RECIST 1.1 and immune-related response criteria (irRC) (Phase II)] and survival were used for efficacy assessments. The treatment was continued until documentation of progressive disease or to a maximum of 12 injections. The trials were performed after approval by a local Human Investigations Committee and in accordance with an assurance filed and approved by the Department of Health and Human Services. This included approval for the long-term follow-up of the melanoma patients analyzed in this review.
Patient population
All eligible patients were treated in the outpatient facility of Mary Crowley Cancer Research (MCCR; Dallas, TX, USA).
IFN-γ ELISPOT assay
The ELISPOT (enzyme-linked immunospot) assay as previously described [17] was performed using the enzyme-linked immunospot assay for IFN-γ, (BD Biosciences, San Jose, CA, USA). Tumor and mononuclear cells were applied on an antibody coated microplate reacting with IFN-γ. Quantitative results in form of reactive spots to IFN-γ, secreted by cytotoxic CD8+ T cells, were measured and used for immune response function analysis. The reading of the ELISPOT plates was performed independently by ZellNet Consulting, Inc. (Fort Lee, NJ, USA). Serial ELISPOT analyses were performed at baseline, Month 2, Month 4 and subsequent time points. Vigil induced ELISPOT conversion (a positive response) was defined as ≥10 spots/105 cells and 2x baseline. All patients were ELISPOT negative at baseline.
Statistical evaluation
Survival was analyzed from time of surgical procurement. Patients were censored for survival on the last known date alive. Analyses of time-to-event variables were performed with the use of log-rank statistics and Kaplan–Meier survival curves. P-values of less than 0.05 were considered to indicate statistical significance. All statistical analyses were performed with the use of IBM SPSS Version 22.
Patient characteristics
A total of 27 patients with advanced melanoma were enrolled in the Phase I and Phase II studies (BB-IND14205: CL-PTL-101, CL-PTL-114). All patients underwent tumor resection as part of the standard care management for palliative control of disease, which also produced tissue for Vigil manufacture. Patient characteristics are shown in table 1. Successful manufacturing of Vigil with 2.5 x 107 cells/ml (Phase I) or 1 x 107 cells/ml (Phase I, Phase II) was achieved in 20 out of 27 patients. The other 7 products could not be released because of insufficient cell dose (n=1) or contamination (n=6). Twelve of the 20 patients received Vigil at 1 x 107 cells/ml dose and all were evaluable for safety and efficacy assessment. The remaining eight were not eligible for treatment for the following reasons: one with ineligible histology and seven with early mortality (<42 days after surgery) prior to planned treatment with Vigil. Thus, 15 patients (7 ineligible, 8 product non-released) who signed consent and underwent surgery for Vigil construction (the intent to treat population; ITT) did not receive Vigil but went on to receive other non-curative standard of care (if available)/experimental treatment. In our previous experience, Vigil release was generally between 21-28 days from manufacture, therefore in order to allow for a conservative assessment, the 3 patients not receiving Vigil who failed to survive 42 days were excluded from the matched comparison (MC) analysis. Thus the conservative MC analysis consists of 12 patients that underwent palliative surgical procedures, had Vigil successfully manufactured and survived ≥42 days.
Table 1. Demographics
|
Vigil |
Intent to Treat |
Matched Comparator |
(n=12) |
(n=15) |
(n=12) |
Age (years) |
|
|
|
Mean |
63.7 |
60.7 |
60.5 |
Range |
32-89 |
39-80 |
49-80 |
Gender |
|
|
|
Female |
6 |
2 |
2 |
Male |
6 |
13 |
10 |
Ethnicity |
|
|
|
Caucasian |
12 |
15 |
12 |
Stage* |
|
|
|
IIIa-c |
3 |
1 |
1 |
IV |
9 |
12 |
10 |
Prior Systemic Therapy |
|
|
|
Chemo |
2 |
4 |
3 |
Radiation |
2 |
3 |
2 |
Checkpoint Inhibitor |
1 |
3 |
1 |
Other (BRAF, investigational) |
4 |
11 |
9 |
Vigil Dose |
|
|
|
1 x 107 cells/ml |
12 |
N/A |
N/A |
N/A: not applicable
All patients required tissue procurement.
*Matched Comparator: F-025 TxNxM0, F-050 T1N0M0. Intent to treat. F-025 TxNxM0
Safety
Nineteen Grade 1 treatment-related adverse events (AE) were observed in the 12 Vigil treated patients. These adverse events were predominately limited to the intradermal injection site in the skin (i.e. local erythema, induration and bruising). There were two Grade 2 treatment-related AEs observed (Table 2). No ≥ Grade 3 AEs related to product were observed.
Table 2. Adverse Events (AEs)
| |
Vigil (n=12) |
Treatment-Related AEs |
Grade 1 |
Grade 2 |
Grade 3 |
Grade 4 |
(n) |
(n) |
(n) |
(n) |
Injection Site – Erythema |
4 |
- |
- |
- |
Injection Site – Induration |
13 |
- |
- |
- |
Rash |
1 |
- |
- |
- |
Probably Treatment-Related AEs |
|
|
|
|
Skin Hypopigmentation |
1 |
- |
- |
- |
Possibly Treatment-Related AEs |
|
|
|
|
Fatigue |
- |
1 |
- |
- |
Irritability |
- |
1 |
- |
- |
Immune response
Using serially obtained PBMCs from each patient, ELISPOT induction/conversion was demonstrated in all patients and, specifically, in 9 of 10 evaluable patients within 3 months of initiation of Vigil. Seven of 10 patients showed an ELISPOT+ response by Month 2, two patients by Month 3 and one patient at the end of treatment (6.5 months after start of treatment) (Figure 1). The ELISPOT+ responses after first dose reflected an increase from 1 spot baseline to a median 78 spots (n=7). Five patients were followed and assessed for ELISPOT reactivity after completion of Vigil and ELISPOT+ responses persisted in all of them (Figure 1). In three, reassessment was limited to two months post treatment completion, but in two repeat ELISPOT reactivity continues beyond 1 year.
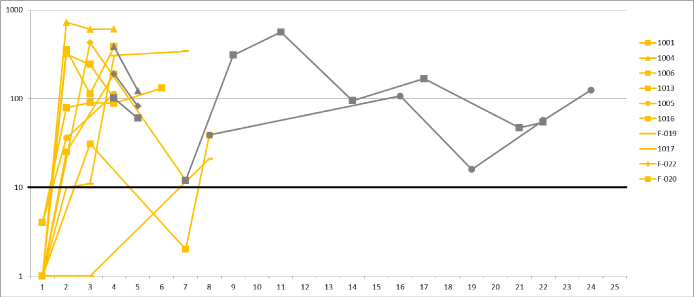
Figure 1. IFN-γ ELISPOT Response to vigil Melanoma ELISPOT + response graph of patients that received Vigil of Phase I and II. Ten patients are represented by two colors i) yellow: on treatment with Vigil and ii) dark gray: off-treatment/follow-up. The y-axis represents the reactive spots on the IFN-γ ELISPOT assay. The x-axis represents different time point of assessments. All patients start out with a negative ELISPOT response status and overcome the threshold of ≥10 spots by Month 3 as the latest. All patients show consistent positive response status at end of treatment with Vigil. Long-term follow-up in two of the patients (F-020, F-022) demonstrate long-term immune response to cancer cells.
Clinical response and survival
Vigil treated patient response is shown in table 3. The median survival from procurement of patients treated with Vigil was 20 months (616 days, range 137-1660 days) compared to both the MC cohort (n=12, not including 3 early mortality patients (<42 days)) who had a median survival of 7 months (208 days; p-value 0.00009) (Figure 2) and the ITT population (n=15 patients) with a median survival of 4 months (122 days). . Eighty-three percent (10/12) of the Vigil treated patients survived ≥1 year from procurement (Table 3).
Table 3. Response of vigil treated patients
Patient ID |
Vigil Cycles Received |
Days Alive Since Procurement |
Days Alive Since Treatment |
Months Since Treatment Start |
Reason for Discontinuation |
Survival Status |
1001 |
4 |
279 |
117 |
3.9 |
Disease Progression |
Dead |
1004 |
4 |
1660 |
1142 |
38.1 |
Normal Completion |
Alive |
1005 |
6 |
498 |
456 |
15.2 |
Disease Progression |
Alive |
1006 |
4 |
1632 |
1156 |
35.5 |
Normal Completion |
Alive |
1008 |
1 |
137 |
11 |
.37 |
Disease Progression |
Dead |
1013 |
4 |
699 |
644 |
21.5 |
Disease Progression |
Dead |
1016 |
7 |
616 |
552 |
18.4 |
Disease Progression |
Alive |
1017 |
8 |
488 |
385 |
12.8 |
Disease Progression |
Dead |
F-005 |
3 |
749 |
560 |
18.7 |
Disease Progression |
Dead |
F-019 |
7 |
572 |
490 |
16.3 |
Normal Completion |
Dead |
F-020 |
7 |
881 |
835 |
27.8 |
Normal Completion |
Dead |
F-022 |
8 |
995 |
942 |
31.4 |
Normal Completion |
Dead |
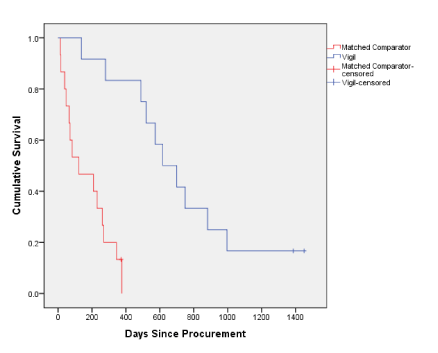
Figure 2. Vigil vs. matched comparator survival since procurement Kaplan Meier Survival Curve of patients with advanced melanoma in Phase I and II of Vigil. The y-axis shows survival rate and the x-axis represents time in days since procurement. The red is the control group (Matched Comparator, n=12) and blue is the Vigil patient cohort (n=12).
Group |
N |
No. of Deaths |
Mean (days) |
Median (days) |
p-value |
Matched Comparator* |
12 |
11 |
204 |
208 |
0.00009 |
Vigil |
12 |
10 |
736 |
616 |
* Excludes 3 patients with survival data <40 days (1009, 1012, F-050) |
This evaluation of Vigil engineered autologous tumor cell therapy in patients with advanced melanoma is preliminary but confirms safety and provides evidence of immune responsiveness (by IFN-γ ELISPOT) in melanoma patients comparable to that previously reported in heavily pretreated patients with other advanced solid tumors and in patients with advanced, recurrent Ewing’s Sarcoma [17,18]. It is encouraging that 8 of the 12 treated patients experienced SD for ≥6 months and that the survival difference was greater than 1 year between the Vigil treated patients and the comparator ITT and MC patient groups. These results are consistent with the long-term follow-up results of Vigil in prior trials, where survival advantage was observed to correlate with ELISPOT activation [19].
2021 Copyright OAT. All rights reserv
Although the MC patient group fulfilled the same inclusion and exclusion criteria as the Vigil treated patients, the gender imbalance, 83:17% vs. 50:50% respectively (Table 1), in this concurrently accrued but non-randomized study, suggests an alternative interpretation of the survival results. In a number of studies, including a pooled analysis of gender as an independent prognostic variable for survival in advanced melanoma [20], gender has been shown to be a significant variable with a female to male survival advantage of approximately 30%. Given the limited number of women in the MC group, in order to address this issue a comparison of survival outcomes was made limited to the men in the Vigil and MC groups. The six Vigil treated men achieved a median survival (dated from procurement) of 657.5 days (range 488-995 days) with a mean of 674.2 days whereas the 10 men in the MC group had a median survival of 238.5 days (range 47-375 days) with a mean of 195 days. Thus, even in a gender specific comparison (within the limits of the data pool from a retrospective combined protocol update), the survival advantage of Vigil over SOC appears to be sustained.
There are several key mechanisms of immune-modulating activity that must be considered for development of effective cancer immunotherapeutics [21]. These include the processing and presentation of cancer related antigens, the specificity of those antigens, antigen presentation through antigen presenting cells (APC, e.g., dendritic cells), MHC peptide/ TCR binding and activation of cytotoxic T cells (CTC), maturation of these T cells into effector and memory subsets, circulation of CTC to target tumor cells, and infiltration into the tumor microenvironment and the recognition of the cancer antigens with consequent cytolysis. Vigil is a unique combinatorial immunotherapeutic that allows for an enhanced immune affector arm by presenting the full panoply of tumor-associated antigens and neoantigens, enhanced activation and attraction of mature dendritic cells by local GM-CSF expression and suppression of TGF-beta related immune suppression, and facilitated acquisition of T cell effector memory function represented by long-term ELISPOT responsiveness post Vigil immunotherapy treatment.
By utilizing the full matrix of patient cancer-related tumor associated antigens (TAAs) and neoantigens the autologous tumor cell Vigil immunotherapy avoids the necessities of epitope identification and HLA matching. Lack of toxic effects in the setting of marked elevation of total body circulating activated T cells (median 1/106 mononuclear cells baseline to 78/106 mononuclear cells post Vigil) suggests that the T cell receptor response was generated to high affinity TAA and neoantigens and, if produced, to below affinity threshold self-antigens. Other approaches such as CAR-T with limited antigen repertoires have thus far shown limited responses in patients with non-hematologic malignancies as well as potentially lethal side effects such as cytokine release syndrome. Vigil, on the other hand, appears to induce a modulated, relevant tumor-related antigen T cell activation that correlates with survival in patients with19 different advanced solid tumor types.
Recent progress in molecular immunology has resulted in the dramatic and oftentimes durable clinical responses seen with immune checkpoint inhibitors in immunogenic melanoma and other supposedly non-immunogenic cancers. The clinical effectiveness of PD-1/PD-L1 axis checkpoint inhibitor therapy (as evidenced by FDA approval in melanoma, NSCLC, renal cell carcinoma, and bladder cancer) indicates that potentially effective tumor-targeting cytotoxic T-lymphocytes (CTLs) are present in the tumor microenvironment, however either 1) are unable to override intrinsic or adaptive resistance, 2) are subject to T cell exhaustion, or 3) are no longer interactive with initially sensitive tumors due to epitope drift and/or evolving somatic mutations and consequent neoantigen formation.
As noted, PD-1/PD-L1 can be either constitutively or inducibly expressed. Further, induction can be either oncogene-driven [22] or T cell-driven (via IFN-γ and STAT3), the latter being the presumptive mechanism of adaptive (tumor cell) resistance [23,24]. There is evidence of vaccine enhanced PD-L1 expression in response to systemic treatment. Similar to Vigil, GVAX, is a GM-CSF producing autologous whole cell tumor vaccine but without intrinsic immunosuppressive TGFβ knockdown. A recent study showed PD-L1 IHC positivity in 12.5% (3 of 25) of resected specimens from unvaccinated patients with pancreatic cancer [25]. Two weeks following GVAX vaccine, specimen membranous PD-L1 expression was increased to 25% (10 of 40) and, in addition, was found in vaccine induced intratumoral tertiary nodules in >80% of patients. In the same report, cyclophosphamide + GVAX (Cy/GVAX) treatment of Panc02 xenografts in C57B16 mice resulted in a 12.5% cure rate compared to 38% with the combination of Cy/GVAX and monoclonal antibody (MAb) targeting PD-1. Likewise the combination significantly increased overall survival (OS) to 81.5 days compared to MAb PD-1 alone, 50 days. Furthermore, in the presence of chronic viral infection or cancer, the persistent exposure of CTLs to high antigen concentrations can result in CD8+ T cell dysfunction; a phenomenon called T cell exhaustion [26]. Treatment with PD-L1/PD-1 axis inhibitors can restore T cell functionality [27]. These data provide a rationale for combining Vigil and an immune checkpoint inhibitor in patients with advanced melanoma and thus provide a basis for both salvage immune checkpoint inhibitor therapy in patients progressing after Vigil, in patients who demonstrate an immune response to Vigil as well as for de novo therapy.
The tumor mutation burden (TMB), not otherwise associated with a survival advantage, has emerged as a potential biomarker for effective PD-L1/PD-1 axis checkpoint inhibitor therapy [28]. Melanoma, in part due to the significant impact of an external mutagen (UV light), is one of the highest TMB expressing cancers. The analysis of immune checkpoint inhibitor responses in patients with heightened mutation rates as a result of preexisting mutations involving specific DNA repair genes; i.e. POLD1, POLE, and DNA mismatch repair (MMR) defects (which play a prominent role in the biogenesis of colorectal cancer [29]). For example, in an analysis of responses to PD-L1 blockade (pembrolizumab), Le and colleagues reported a 78% immune related PFS for MMR deficient patients versus 11% in MMR proficient patients [30]. In addition, there was a 40% PR vs. 0% PR, in the two groups, respectively. Rizvi et al addressed the underlying mechanism by hypothesizing (as others have) that recognition of neoantigens, formed as a consequence of somatic mutations (particularly missense and frameshift), is important for the activity of anti PD-1 therapy. They then characterized the neoantigen tumor landscape in these same patients and found a direct correlation with TMB (p < 0.0001). Cancers (regardless of histology) with a mean mutational load of >10 somatic mutations per Mb of coding DNA are likely to have enough cells capable of proteasome processing and adequate MHC I:peptide binding affinity to produce epitopes recognizable by T cells [31,32]. However, insofar as these neoantigenic epitopes elicit antitumor immune responses, they also have the potential to induce off-setting counter responses including CTLA4, PD-1, and PD-L1 [33] thereby accounting, at least in part, for the benefit derived from checkpoint inhibitors.
In conclusion, given 1) the apparent effectiveness of the engineered autologous tumor cell Vigil immunotherapy, 2) oncogene or immunotherapy mediated IFNγ-induced expression of the PD-1/PD-L1 axis components (adaptive resistance), 3) the enhanced effectiveness of GM-CSF secreting autologous tumor cell therapies combined with anti-PD-1/PD-L1 axis MAb, 4) PD-1/PD-L1 blockade reversion of T cell exhaustion [34,35], and 5) the limited response activity to monomodal anti-PD-1/PD-L1 in PD-L1 negative populations, it is our contention that Vigil immunotherapy upregulation of activated T cell populations, as a result of combining both local GMCSF related immune enhancement with down-regulation of intrinsic tumor cell immunosuppressive TGFβ, Vigil will produce an additive if not synergistic combinatorial immunotherapeutic regimen in conjunction with immune checkpoint inhibition. Such a study is in progress.
We gratefully acknowledge the generous support of the Jasper L. and Jack Denton Wilson Foundation, Joe and Jessie Crump Foundation Medical Research Fund, MMK Foundation, Redman Foundation and Summerfield G. Roberts Foundation. In honor of Joseph Kuhn, MD, who passed away from cancer before this work could be completed and who was a good friend and colleague of all authors, as well as, significant contributor to this work, his name is listed with permission of his wife, Mollie Kuhn. The authors would like to acknowledge Michelle Watkins and Brenda Marr for their competent and knowledgeable assistance in the preparation of the manuscript.
The following authors are shareholders in Gradalis, Inc. and Strike Bio: Jeffrey Lamont, Padmasini Kumar, Gladice Wallraven, Neil Senzer and John Nemunaitis. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
- Ma MW, Medicherla RC, Qian M, Vega-Saenz de Miera E, Friedman EB, et al. (2012) Immune response in melanoma: an in-depth analysis of the primary tumor and corresponding sentinel lymph node. Mod Pathol 25: 1000-1010. [Crossref]
- Ribero S, Osella-Abate S, Sanlorenzo M, Savoia P, Astrua C, et al. (2013) Favourable prognostic role of regression of primary melanoma in AJCC stage I-II patients. Br J Dermatol 169: 1240-1245. [Crossref]
- Ribero S, Gualano MR, Osella-Abate S, Scaioli G, Bert F, et al. (2015) Association of Histologic Regression in Primary Melanoma With Sentinel Lymph Node Status: A Systematic Review and Meta-analysis. JAMA Dermatol 151: 1301-1307. [Crossref]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214-218. [Crossref]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711-723. [Crossref]
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, et al. (2015) Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol 16: 522-530.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, et al. (2016) Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival analysis of KEYNOTE-006. J Clin Oncol 34: 9504.
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, et al. (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16: 375-84.
- Larkin J1, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, et al. (2015) Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373: 23-34. [Crossref]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, et al. (2015) Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 372: 2521-2532. [Crossref]
- Robert C1, Long GV, Brady B, Dutriaux C, Maio M, et al. (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372: 320-330. [Crossref]
- Senzer NN1, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, et al. (2009) Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27: 5763-5771. [Crossref]
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, et al. (2015) Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J Clin Oncol 33: 2780-2788. [Crossref]
- Andtbacka RH, Agarwala SS, Ollila DW, Hallmeyer S, Milhem M, et al. (2016) Cutaneous head and neck melanoma in OPTiM, a randomized phase 3 trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor for the treatment of unresected stage IIIB/IIIC/IV melanoma. Head Neck. [Crossref]
- Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, et al. (2012) Phase I trial of "bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell" vaccine (FANG) in advanced cancer. Mol Ther 20: 679-686. [Crossref]
- Maples PB, Kumar P, Yu Y, Wang Z, Jay C, et al. (2010) FANG Vaccine: Autologous Tumor Vaccine Genetically Modified to Express GM-CSF and Block Production of Furin. BioProcessing Journal 8: 4-14.
- Nemunaitis J, Barve M, Orr D, Kuhn J, Magee M, et al. (2014) Summary of bi-shRNA/GM-CSF augmented autologous tumor cell immunotherapy (FANGTM) in advanced cancer of the liver. Oncology 87: 21-29. [Crossref]
- Ghisoli M, Barve M, Mennel R, Lenarsky C, Horvath S, et al. (2016) Three-year Follow up of GMCSF/bi-shRNA(furin) DNA-transfected Autologous Tumor Immunotherapy (Vigil) in Metastatic Advanced Ewing's Sarcoma. Mol Ther 24: 1478-1483. [Crossref]
- Nemunaitis J, Barve M, Orr D, Kuhn J, Magee M, et al. (2014) Summary of bi-shRNA/GM-CSF augmented autologous tumor cell immunotherapy (FANGTM) in advanced cancer of the liver. Oncology 87: 21-29. [Crossref]
- Joosse A, Collette S, Suciu S, Nijsten T, Patel PM, et al. (2013) Sex is an independent prognostic indicator for survival and relapse/progression-free survival in metastasized stage III to IV melanoma: a pooled analysis of five European organisation for research and treatment of cancer randomized controlled trials. J Clin Oncol 31: 2337-2346. [Crossref]
- Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39: 1-10. [Crossref]
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, et al. (2013) Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 3: 1355-1363. [Crossref]
- Yao S, Chen L (2013) Adaptive resistance: a tumor strategy to evade immune attack. Eur J Immunol 43: 576-579. [Crossref]
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, et al. (2012) Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 4: p. 127ra37. [Crossref]
- Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, et al. (2015) PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother 38: 1-11. [Crossref]
- Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, et al. (2006) PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol 80: 11398-11403. [Crossref]
- Zarour HM (2016) Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res 22: 1856-1864. [Crossref]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, et al. (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348: 124-128. [Crossref]
- Timmermann B, Kerick M, Roehr C, Fischer A, Isau M, et al. (2010) Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One 5: e15661. [Crossref]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, et al. (2015) PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372: 2509-2520. [Crossref]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, et al. (2013) Signatures of mutational processes in human cancer. Nature 500: 415-421. [Crossref]
- Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348: 69-74. [Crossref]
- Matsushita H, Sato Y, Karasaki T, Nakagawa T, Kume H, et al. (2016) Neoantigen Load, Antigen Presentation Machinery, and Immune Signatures Determine Prognosis in Clear Cell Renal Cell Carcinoma. Cancer Immunol Res 4: 463-471 [Crossref]
- Yao S, Chen L (2006) Reviving exhausted T lymphocytes during chronic virus infection by B7-H1 blockade. Trends Mol Med 12: 244-246. [Crossref]
- Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH (2006) Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med 203: 2223-2227. [Crossref]