Abstract
The inter-individual variability in drug metabolism is considered as one of the most important factors that influence drug response i.e. efficacy and toxicity. This variability in drug response could be due to inhibition of the metabolic enzymes or transporters of drug elimination, co-administration of other drugs or the genetic variation in the metabolizer or transporter genes. When a given genetic variant is found in a certain population with a frequency of more than 1%, it is defined as polymorphic variant that results in an altered protein function. Based on the genetic polymorphism of drug metabolizing enzymes, a given population may be classified into three groups viz., (i) extensive metabolizers, carrying normal-function metabolic enzymes wherein levels of drug efficacy and drug elimination are balanced, (ii) ultra-extensive metabolizers, having extraordinary-function metabolic enzymes wherein drug elimination is enhanced, and (iii) poor-metabolizers with less active or no metabolic enzymes wherein drug accumulation is likely to occur due to reduced elimination. In the recent decade, several comprehensive studies have been conducted to investigate the genetic polymorphism of the metabolic enzymes and transporters in different ethnic groups. Such studies have enhanced our understanding of the mechanisms of drug toxicity in a given population as well as the ability to predict individuals who are predisposed to drug toxicity, reduced efficacy or environmental exposure-linked diseases. The present article reviews and discusses the genetic variants of genes that are heavily involved in drug elimination and transport.
Introduction
In general, prior to approval for the clinical use of a drug, extensive studies or trials have to be done to evaluate its efficacy and safety. The safety of drug treatment is a major concern since the toxicity and adverse drug reaction (ADR) have been identified as a significant factor in patient mortality. ADR can be defined as ‘an appreciably harmful or unpleasant reaction, resulting from an intervention related to the use of a medical product, which predicts hazard from future administration and warrants prevention or specific treatment, or alteration of the dosage regiment, or withdrawal of the product’ [1]. In the United States, drug toxicity and ADR occur in 1 out of 15 patients and represent the fourth to sixth leading cause of approximately 140,000 fatalities per year [2,3]. This forced the withdrawal of 10% of approved drugs from the market between 1975-1999 [4]. The economic burden resulting from drug-related morbidity and mortality in the US alone exceeded $177.4 billion in year 2000 [5]. Therefore, a better understanding of the underlying mechanism of ADR is necessary to prevent the significant outcomes of such a major health concern.
The pharmacologic or toxicologial effect of a drug is related to the persisting level of the drug within the body where any modification in that level might alter body’s biochemical and/or physiological homeostasis. Any change in the processes of absorption, metabolism, distribution, or excretion affects the pharmacokinetic of drugs and their effects (Figure 1). Several polymorphisms that have been linked to variations in drug response involve genes that code for drug metabolizing enzymes and transporters.
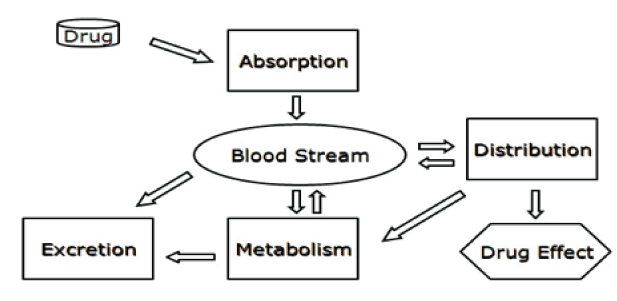
Figure 1. The pathways most of drugs undergo in order to express their activities
Drug metabolism is an essential process by which human body gets rid of foreign lipophilic molecules usually via their conversion to more hydrophilic molecules that can be easily eliminated. Drug metabolism can be divided into two phases: phase-I (functionalization reactions) in which certain metabolic enzymes (mainly cytochrome P450 enzymes) metabolize drugs mainly via oxidation and to a lesser extent via reduction, hydrolysis and hydration, and Phase-II (conjugation reactions) in which sets of enzymes, such as UDP glucuronosyltransferases (UGTs), N-acetyl transferases (NATs), and esterases metabolize drugs via addition of large endogenous hydrophilic moieties (Figure 2).
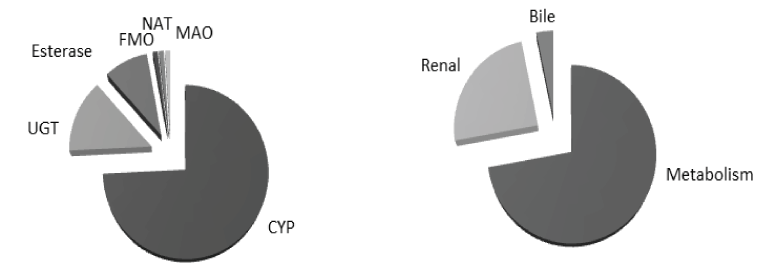
Figure 2. Ilustration of pathways involved in drug elimination (left) and contribution of metabolic enzymes in drug metabolism (right) [6]
Beside these metabolic enzymes, it has been found that transporter enzymes, such as the organic anion transporter which control drugs excretion also play crucial role in determining drugs activities and toxicities. Production of these enzymes is highly regulated by a set of nuclear receptors such as pregnane X. Although levels of enzymes or proteins in human body can vary from one to another due to environmental factors, genetic variation also has a major impact.
Cytochrome P450 (CYP) enzyme
CYP are heme-containing, membrane-bound, and endoplasmic reticulum-located proteins [7,8] that catalyze the initial step in the oxidative metabolism of a plethora of endogenous (steroids, bile acids, fatty acids, prostaglandins, leukotrienes, and biogenic amines) and exogenous (drugs, carcinogens, dietary supplements, pollutants, pesticides, and environmental chemicals) substances [9-14]. The name was derived from the cytochrome pigment (P) having a 450 nm ultraviolet spectral peak when reduced and bound to carbon monoxide [15-18].
Based on similarities in their protein sequences, CYP enzymes have been divided into families and subfamilies [10,19]. Enzymes with ≤40% sequence similarity are grouped into different families, designated by an Arabic number (e.g. CYP2). The enzymes with 40-55% similarity are grouped into different subfamilies, designated by an English alphabet (e.g. CYP2C). Enzymes with ≥55% similarity are classified as members of the same subfamily, and suffixed with Arabic number (e.g. CYP2C9). So far, fifty seven functional CYP genes and eighteen families have been identified in humans, among them only the first three families are involved in drug metabolism [15,20-23]. Of these CYP3A4 followed by 2D6, 2C9, and 2C19 contribute predominantly in metabolism of commonly used drugs (Figure 3).
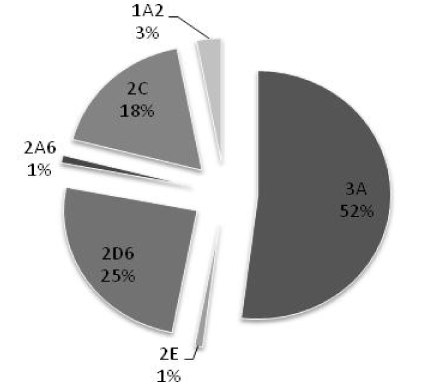
Figure 3. Contribution of CYP450 in drug metabolism [6]
The human CYP1A subfamily consists of two members, CYP1A1 and CYP1A2, which are located in chromosome 15 [24]. CYP1A2 is expressed almost exclusively in the liver, although low expression has also been detected in the lungs and intestines. CYP1A is responsible for activation of nitrosamines, arylamines, polycyclic aromatic amines (PAHs), and aflatoxin B1 into intermediates that can bind DNA and induce mutation [25-28]. It has been speculated that 90% of all known pro-carcinogens are activated by CYP1A1 and CYP1A2 [29]. CYP1A2 is responsible for metabolism of some food supplements and drugs such as caffeine and theophylline [30,31], and is highly inducible by nicotine, charbroiled foods, and cruciferous vegetables [32,33].
The human CYP2C subfamily consists of four genes, CYP2C8, CYP2C9, CYP2C18, and CYP2C19, which are located in chromosome 10 [34]. CYP2C9 is the most abundant CYP2C protein expressed in the liver. Lower levels of CYP2C9 expression have been detected in the kidneys and intestines. CYP2C9 is responsible for metabolizing many drugs such as tolbutamide and S-warfarin [35,36]. Its expression is subjected to induction by many drugs including phenobarbital and rifampicin [37,38].
Within the CYP2D subfamily in humans, CYP2D6 is the only active gene which is located in chromosome 22 [39]. CYP2D6 is expressed in the liver and to lesser extent in the intestines and brain. It is responsible for metabolizing a wide variety of prescribed drugs such as dextromethorphan and debrisoquine [40]. The CYP2D6 gene is characterized by its high genetic polymorphism and its resistance to induction [41,42].
The human CYP2E subfamily contains a single gene, CYP2E1, which is located in chromosome 10 [43]. CYP2E1 is expressed mostly in the liver and to a lesser extent in the kidneys and lungs. It is responsible for metabolism of many compounds including ethanol and chlorzoxazone [44,45]. CYP2E1 expression is subject to induction by a variety of compounds such as ethanol and isoniazid [45-47].
The human CYP3A subfamily consists of four genes, CYP3A4, CYP3A5, CYP3A7, and CYP3A43, which are located in chromosome 7 [48]. CYP3A4 is highly expressed in the liver and intestines and to a lesser extent in the lungs. CYP3A4 is responsible for metabolism of a wide variety of drugs including nifedipine and erythromycin [49,50] and activation of many carcinogens such as polycyclic hydrocarbons (PAH) and aflatoxin B1 [25,27]. CYP3A4 is the most highly inducible CYP gene, and numerous pharmaceutical compounds, including rifampicin and dexamethasone, are able to enhance the expression of this gene [51,52].
UDP glucuronosyltransferase (UGT) enzyme
Conjugation with glucuronic acid is responsible for the elimination of a diverse range of xenobiotics and endogenous compounds in humans and other mammalian species. Glucuronidation serves as a clearance mechanism for drugs from almost all therapeutic classes, including dietary chemicals, environmental pollutants and chemical carcinogens, and phase-I oxidation products. Endogenous compounds metabolized by glucuronidation include bile acids, bilirubin, hydroxy- steroids and thyroid hormones. Although a number of bioactive glucuronides are known, glucuronidation is primarily considered a detoxification process. Glucuronidation reactions are catalyzed by UGT, the microsomal enzyme with a broad substrate profile. UGT genes have been classified into families and subfamilies based on evolutionary divergence, with all known UGT being included in the 1A, 2A and 2B subfamilies [53-55]. UGT family 1 isoforms are derived from a single gene locus which spans more than 500 kb on chromosome 2q37. UGT1A comprises at least 12 promoters and first exons, which are spliced separately to common exons 2-5 resulting in transcripts which encode enzymes with unique amino termini preceding an identical carboxyl terminus of 245 amino acids. The UGT1A locus encodes nine functional enzymes; 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9 and 1A10 [54,56]. UGT family 2 enzymes include 2A1, 2B4, 2B7, 2B10, 2B11, 2B15 and 2B17. Among all these enzymes UGT2B7 followed by UGT1A1 contributes predominantly in drug metabolism [57].
Drug transporter proteins
Many prescribed medications or their metabolites exist as organic anions at physiological pH. These compounds are ultimately handled by the organic anion transport systems of the kidney, liver, and choroid plexus, perhaps the best studied of which is the ‘classic’ multispecific organic anion secretory pathway of the renal proximal tubule. The organic anion transporter (OAT) comprises OAT1 (slc22a6), the prototypical OAT, OAT2 (slc22a7), OAT3 (slc22a8), OAT4 (slc22a11), and OAT5. Substrates of this secretory pathway are strikingly diverse and include such clinically important pharmaceuticals as beta-lactam antibiotics, probenecid, loop and thiazide diuretics, angiotensin converting enzyme (ACE) inhibitors, nonsteroidal anti-inflammatory drugs (NSAID), and methotrexate, among others, as well as various endogenous compounds, including cyclic nucleotides, prostaglandins, folate, neurotransmitter-metabolites, and hormone-conjugates [58].
The ATP binding cassette (ABC) is a ubiquitous and important family of such transporter proteins. Members of this super family are present in mammals as well as in prokaryotic organisms and use ATP as the energy source to activate the extrusion process. P-glycoprotein (P-gp) is the most important and widely studied member of ABC super family. P-gp is anatomical localized in several normal human tissues with excretory (liver, kidney, adrenal gland) and barrier (intestine, blood-brain barrier, placenta, blood-testis and blood-ovarian barriers) functions. Recently, it has been found that P-gp has an important physiological role in detoxification and protection of the body against toxic xenobiotics and metabolites. Its detoxification role is carried out by secreting toxic compounds into bile, urine, and the intestinal lumen and by preventing their accumulation in human body. Additionally, clinically impotant ADR were reported when digoxin (a good P-gp substrate) used with other drugs, such as quinidine, verapamil, talinolol, clarithromycin, itraconazole, erythromycin, and propafenone [59].
Multidrug and toxin extrusion (MATE) proteins (METAE1, MATE2, MATE2-K and MATE2-B) are the recently identified transporter that mediates the final excretion step for organic cations [60,61]. The MATE-mediated transport is driven by oppositely directed proton gradient, and are therefore, considered secondary active transporters. MATE proteins are involved in physiological and/or pharmacological processes such as pharmacokinetics, resistance in tumor tissues, and hormone secretion. Because of its importance, drug regulatory authorities are considering incorporation of MATE enzymes into their guidelines.
The Level of any given protein is primarily regulated via controlled gene expression, a process in which the gene produces protein via transcription followed by translation. Modulation (inhibition or induction) of the activity of a given protein could be due to direct effect on the protein its modulator or through alteration in gene expression [62]. For instance, it has been found that the induction of CYP450 enzymes was attributed to elevation in the transcriptional activity of CYP genes [63,64]. Many CYP subfamilies including CYP1A, CYP2B, CYP2C, and CYP3A, are highly inducible by xenobiotics [21]. Therefore, the outcome of enzyme induction depends on the pharmacological activity of the parent compounds and their metabolites.
When the parent compound is the active therapeutic agent, then the net effect of enzyme induction could be loss of the pharmacological efficacy. For example, rifampicin increases the CYP3A4-dependent metabolism of cyclosporine resulting in rejection of the transplanted organ by the body [65]. On the other hand, when the metabolite is more active than the parent compound or toxic, then the induction will increase the chance for toxicity. For example, ethanol increases the CYP2E1-dependent metabolism of acetaminophen resulting in formation of its hepatotoxic metabolite (N-acetyl-p-benzoquinoneimine) [66]. The examples mentioned as well as many others all reveal the clinical consequences of Drug elimination enzymes induction and suggest that more studies are required in order to fully understand the mechanisms underlying their regulation.
In the recent decade, extensive efforts have been made to understand the molecular mechanisms underlying the expression of CYP enzymes. It has been found that the induction of CYP enzymes is primarily regulated by a group of orphan nuclear receptors. They are called orphans because they were identified without knowing their endogenous or exogenous ligands [67]. These receptors share two essential functional domains that include the N-terminal DNA-binding domain (DBD) and the C-terminal ligand-binding domain (LBD) [68]. The conserved DBD acts to link the receptor to specific 5’-flanking region (5’-FR) element in its target gene called the xenobiotic responsive element (XRE) [67]. The less conserved LBD has at least four functions: ligand binding, binding of co-activators or co-repressors, dimerization, and transactivation [69]. Nuclear receptors were considered prime candidates for mediating hepatic drug induction for several reasons [21]. First, their ligands are small and lipophilic similar to those of CYP enzymes. They bind to specific DNA elements similar to those found in the 5’-flanking sequences (5’-FSs) of CYP genes. Furthermore, they are expressed in specific tissues where most CYP enzymes are expressed. Finally, they play key roles in many physiological processes in which P450 enzymes are involved.
The most studied nuclear orphan receptor is the pregnane X receptor (PXR). PXR was isolated and identified as a key regulator in CYP3A expression in 1998 [70-72], although recent studies have disclosed its regulatory role for other CYP genes such as CYP2C and CYP2B [73,74]. PXR is expressed predominantly in the liver and intestines and to a lesser extent in the kidneys and lungs [75]. Many chemicals including prescription drugs, steroids, and environmental factors are able to bind and activate PXR [70,76]. For example, the antibiotic rifampicin and the antidepressant herbal product hyperforin are potent PXR activators [77,78]. The biochemical process of PXR activation has been illustrated [68]. Upon ligand binding, a conformational change in the LBD creates a co-activator (e.g. steroid receptor coactivator-1, SRC-1) binding surface; and transcriptional activation occurs after recruitment of co-activator to the receptor [69]. Subsequently, PXR regulates gene expression by forming a heterodimer with the retinoid X receptor (RXRα) and then regulation is achieved by binding of the PXR-RXRα heterodimer to XRE present in the 5’-FR of the target gene (Figure 4). The unique feature of PXR-mediated induction is its species specificity, primarily due to the differences in LBD [70]. For example, rifampicin is a potent activator for human PXR but not the rodent isoform, whereas pregnenolone-16α-carbonitrile (PCN), an anti-glucocorticoid, is a rodent-specific activator. PXR humanized mice have been generated [79] and in these transgenic mice the profile of PXR-based induction was similar to the human profile.
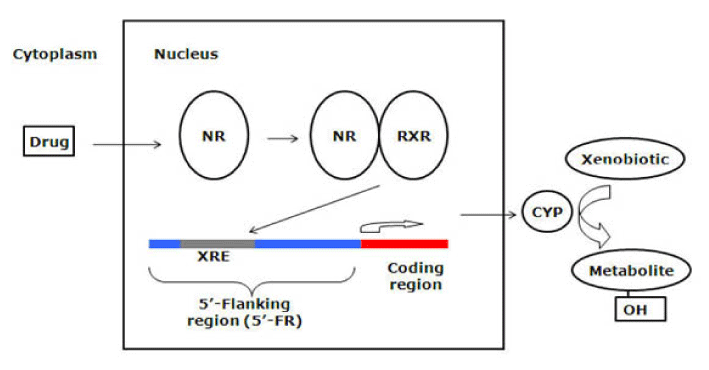
Figure 4. Mechanism of nuclear receptor (NR) activation by drugs and their control of metabolic enzymes and transporters
Another important nuclear receptor is the constitutive androstane receptor (CAR). CAR was first isolated in 1994, but its role in CYP2B induction was not appreciated until 1998 [80-82]. CAR also can regulate other CYP genes such as CYP3A and CYP2C [83,84]. CAR is expressed predominantly in the liver and intestines and can be activated by many drugs such as phenobarbital and phenytoin. The mechanism of CAR activation is more complex than that of PXR. CAR is cytosolic protein and upon activation, it translocates into the nucleus and forms a heterodimer with RXRα. Similar to PXR, it is the heterodimer that binds to target gene sequence and activates transcription [85-87]. Phenobarbital activates CAR by facilitating its nuclear translocation through a phosphorylation-based mechanism [85,87,88]. The only molecules shown to directly bind CAR were androstanol and clotrimazole which are inverse agonists that deactivate the response [89]. Like PXR, CAR shows species differences in its induction profile, for example 1,4-bis[2-(3,5 dichloropyridyloxy)]benzene (TCPOBOP) was found to be specific mouse CAR activator [88,90]. The broad role of CAR and PXR in regulating many metabolizing enzymes and transporters and cross-regulation of gene expression has been reported [91].
Perhaps the most well studied nuclear receptor is the aryl-hydrocarbon receptor (AhR). For more than thirty years, AhR has been known to be a CYP1A regulator [91-94]. AhR is a helix-loop-helix protein that belongs to the polycyclic aromatic hydrocarbon (PAH) family of transcription factors. Similar to CAR, AhR is a cytosolic protein and becomes activated once activated by its ligand. Consequently, the activated receptor translocates into the nucleus, forms a heterodimer with its nuclear translocator protein (ARNT), binds to XRE sequences upstream of CYP1 genes, and activates gene transcription [95,96]. AhR-dependent induction is conserved among many cell types and across animal species. A significant number of substances were found to be ligands for AhR including omeprazole as well as several important environmental carcinogens found in auto exhaust and cigarette smoke [97,98].
Other nuclear receptors are also known to involve in CYP regulation. For example, the peroxisome proliferator-activated receptor (PPARα) regulates CYP4A [99-101] and the vitamin D receptor (VDR) regulates CYP3A, CYP2B, CYP2C, and CYP24 [102-103]. The liver X receptor (LXR) and the farnesol X receptor (FXR) both regulate the expression of CYP7A [104-107]. Additionally, some transcriptional factors play crucial role in CYP regulation. For example, the hepatic nuclear factor (HNF1α) regulates the expression of CYP2E1, CYP1A2, CYP7A1, and CYP27 [108] and the HNF4α regulates CYP3A, CYP2C, CYP2D6, CYP2A6, and CYP2B [109-111]. Other transcriptional factors such as the HNF3γ regulate CYP2C [112] while the CAAT/enhancer binding protein (C/EBP) regulates the expression of CYP2B, CYP2D, and CYP2C [113]. Similar to CYP450 genes, expression levels of P-gp, OAT, and of course UGTs were found to be greatly regulated by nuclear receptors, the pregnane X receptor (PXR) particularly [114,115].
The mammalian MATE 1 is encoded by SLC47A1 gene. Polymorphisms in SLC47A genes may affect renal excretion of substrate drugs, such as metformin, resulting in inadequate pharmacotherapy or occurrence of toxic effects [116]. However, expression and function of MATE enzymes in tissues other than kidney and liver remain to be elucidated.
Conclusion
The efficacy as well as safety of therapeutic drugs is a major concern in patient management where inter-individual genetic variability in drug metabolizer and transporter genes greatly influences drug response. Ample of studies on molecular mechanisms of such the genetic polymorphism have enhanced our understanding of drug metabolism, and to predict its toxicity in a given population. Nevertheless, a comprehensive and better understanding of the drug toxicity is necessary to prevent the significant outcomes of such a major health concern.
References
- Edwards IR, Aronson JK (2000) Adverse drug reactions: definitions, diagnosis, and management. Lancet 356: 1255-1259. [Crossref]
- Davis MP, Homsi J (2001) The importance of cytochrome P450 monooxygenase CYP2D6 in palliative medicine. Support Care Cancer 9: 442-451. [Crossref]
- Johnson MD1, Newkirk G, White JR Jr (1999) Clinically significant drug interactions. Postgrad Med 105: 193-195, 200, 205-6 passim. [Crossref]
- Severino G, Del Zompo M (2004) Adverse drug reactions: role of pharmacogenomics. Pharmacol Res 49: 363-373. [Crossref]
- Ernst FR, Grizzle AJ (2001) Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 41: 192-199. [Crossref]
- Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, et al. (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 32: 1201-1208. [Crossref]
- Liddle C, Goodwin B (2002) Regulation of hepatic drug metabolism: role of the nuclear receptors PXR and CAR. Semin Liver Dis 22: 115-122. [Crossref]
- Tucker GT (2000) Advances in understanding drug metabolism and its contribution to variability in patient response. Ther Drug Monit 22: 110-113. [Crossref]
- Omura T (1999) Forty years of cytochrome P450. Biochem Biophys Res Commun 266: 690-698. [Crossref]
- Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, et al. (1996) P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6: 1-42. [Crossref]
- Gonzalez FJ (1988) The molecular biology of cytochrome P450s. Pharmacol Rev 40: 243-288. [Crossref]
- Guengerich FP (1989) Characterization of human microsomal cytochrome P-450 enzymes. Annu Rev Pharmacol Toxicol 29: 241-264.
- Waxman DJ, Azaroff L (1992) Phenobarbital induction of cytochrome P-450 gene expression. Biochem J 281 : 577-592. [Crossref]
- Gonzalez FJ (1990) Molecular genetics of the P-450 superfamily. Pharmacol Ther 45: 1-38. [Crossref]
- Nebert DW, Russell DW (2002) Clinical importance of the cytochromes P450. Lancet 360: 1155-1162. [Crossref]
- Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. i. evidence for its hemoprotein nature. J Biol Chem 239: 2370-2378. [Crossref]
- Klingenberg M (1958) Pigments of rat liver microsomes. Arch Biochem Biophys 75: 376-386. [Crossref]
- Estabrook RW, Hildebrandt AG, Baron J, Netter KJ, Leibman K (1971) A new spectral intermediate associated with cytochrome P-450 function in liver microsomes. Biochem Biophys Res Commun 42: 132-139.
- Nelson DR, Kamataki T, Waxman DJ, et al. (1993). The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes, and nomenclature. DNA Cell Biol 12: 1-51. [Crossref]
- Waxman DJ (1999) P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys 369: 11-23. [Crossref]
- Handschin C, Meyer UA (2003) Induction of drug metabolism: the role of nuclear receptors. Pharmacol Rev 55: 649-673. [Crossref]
- Pirmohamed M, Park BK (2003) Cytochrome P450 enzyme polymorphisms and adverse drug reactions. Toxicology 192: 23-32. [Crossref]
- Ingelman-Sundberg M (2002) Polymorphism of cytochrome P450 and xenobiotic toxicity. Toxicology 181-182: 447-52. [Crossref]
- Hildebrand CE, Gonzalez FJ, McBride OW, Nebert DW (1985) Assignment of the human 2,3,7,8-tetrachlorodibenzo-p-dioxin-inducible cytochrome P1-450 gene to chromosome 15. Nucleic Acids Res 13: 2009-2016. [Crossref]
- Aoyama T, Yamano S, Guzelian PS, Gelboin HV, Gonzalez FJ (1990) Five of 12 forms of vaccinia virus-expressed human hepatic cytochrome P450 metabolically activate aflatoxin B1. Proc Natl Acad Sci U S A 87: 4790-4793. [Crossref]
- Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, et al. (1997). Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis 18: 851-854. [Crossref]
- Hecht SS (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 11: 559-603. [Crossref]
- Wakabayashi K1, Nagao M, Esumi H, Sugimura T (1992) Food-derived mutagens and carcinogens. Cancer Res 52: 2092s-2098s. [Crossref]
- Rendic S, Di Carlo FJ (1997) Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev 29: 413-580. [Crossref]
- Miners JO, Birkett DJ (1996) The use of caffeine as a metabolic probe for human drug metabolizing enzymes. Gen Pharmacol 27: 245-249. [Crossref]
- Miller M, Opheim KE, Raisys VA, Motulsky AG (1984) Theophylline metabolism: variation and genetics. Clin Pharmacol Ther 35: 170-182. [Crossref]
- Sesardic D, Boobis AR, Edwards RJ, Davies DS (1988) A form of cytochrome P450 in man, orthologous to form d in the rat, catalyses the O-deethylation of phenacetin and is inducible by cigarette smoking. Br J Clin Pharmacol 26: 363-372. [Crossref]
- Guengerich FP (1994) Catalytic selectivity of human cytochrome P450 enzymes: relevance to drug metabolism and toxicity. Toxicol Lett 70: 133-138. [Crossref]
- Gray IC, Nobile C, Muresu R, Ford S, Spurr NK (1995) A 2.4-megabase physical map spanning the CYP2C gene cluster on chromosome 10q24. Genomics 28: 328-332. [Crossref]
- Miners JO1, Birkett DJ (1996) Use of tolbutamide as a substrate probe for human hepatic cytochrome P450 2C9. Methods Enzymol 272: 139-145. [Crossref]
- Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, et al. (1992) Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol 5: 54-59. [Crossref]
- Heimark LD, Gibaldi M, Trager WF, O'Reilly RA, Goulart DA (1987) The mechanism of the warfarin-rifampin drug interaction in humans. Clin Pharmacol Ther 42: 388-394. [Crossref]
- Breckenridge A, Orme M (1971) Clinical implications of enzyme induction. Ann N Y Acad Sci 179: 421-431. [Crossref]
- Gonzalez FJ, Vilbois F, Hardwick JP, McBride OW, Nebert DW, et al. (1988) Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Genomics 2: 174-179. [Crossref]
- Henthorn TK, Benitez J, Avram MJ, Martinez C, Llerena A, et al. (1989) Assessment of the debrisoquin and dextromethorphan phenotyping tests by gaussian mixture distributions analysis. Clin Pharmacol Ther 45: 328-333. [Crossref]
- Mahgoub A, Idle JR, Dring LG, Lancaster R, Smith RL (1977) Polymorphic hydroxylation of Debrisoquine in man. Lancet 2: 584-586. [Crossref]
- Eichelbaum M, Mineshita S, Ohnhaus EE, Zekorn C (1986) The influence of enzyme induction on polymorphic sparteine oxidation. Br J Clin Pharmacol 22: 49-53. [Crossref]
- Kölble K (1993) Regional mapping of short tandem repeats on human chromosome 10: cytochrome P450 gene CYP2E, D10S196, D10S220, and D10S225. Genomics 18: 702-704. [Crossref]
- Girre C, Lucas D, Hispard E, Menez C, Dally S, et al. (1994) Assessment of cytochrome P4502E1 induction in alcoholic patients by chlorzoxazone pharmacokinetics. Biochem Pharmacol 47: 1503-1508. [Crossref]
- Lieber CS (1997) Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev 77: 517-544. [Crossref]
- Koop DR, Tierney DJ (1990) Multiple mechanisms in the regulation of ethanol-inducible cytochrome P450IIE1. Bioessays 12: 429-435. [Crossref]
- Gonzalez FJ, Ueno T, Umeno M, Song BJ, Veech RL, et al. (1991) Microsomal ethanol oxidizing system: transcriptional and posttranscriptional regulation of cytochrome P450, CYP2E1. Alcohol Alcohol Suppl 1: 97-101. [Crossref]
- Inoue K, Inazawa J, Nakagawa H, Shimada T, Yamazaki H, et al. (1992) Assignment of the human cytochrome P-450 nifedipine oxidase gene (CYP3A4) to chromosome 7 at band q22.1 by fluorescence in situ hybridization. Jpn J Hum Genet 37: 133-138. [Crossref]
- Breimer DD, Schellens JH, Soons PA (1989) Nifedipine: variability in its kinetics and metabolism in man. Pharmacol Ther 44: 445-454. [Crossref]
- Watkins PB, Murray SA, Winkelman LG, Heuman DM, Wrighton SA, et al. (1989) Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. Studies in rats and patients. J Clin Invest 83: 688-697. [Crossref]
- Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB (1992) Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest 90: 1871-1878. [Crossref]
- Schuetz EG, Guzelian PS (1984) Induction of cytochrome P-450 by glucocorticoids in rat liver. II. Evidence that glucocorticoids regulate induction of cytochrome P-450 by a nonclassical receptor mechanism. J Biol Chem 259: 2007-2012. [Crossref]
2021 Copyright OAT. All rights reserv
- Ritter JK (2000) Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem Biol Interact 129: 171-193. [Crossref]
- Mackenzie PI, Owens IS, Burchell B, Bock KW, Bairoch A, et al. (1997). The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics 7: 255-269. [Crossref]
- Radominska-Pandya A, Czernik PJ, Little JM, Battaglia E, Mackenzie PI (1999) Structural and functional studies of UDP-glucuronosyltransferases. Drug Metab Rev 31: 817-899. [Crossref]
- Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, et al. (2000). Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat 16: 297-306. [Crossref]
- Gregory PA, Lewinsky RH, Gardner-Stephen DA, Mackenzie PI (2004) Regulation of UDP glucuronosyltransferases in the gastrointestinal tract. Toxicol Appl Pharmacol 199: 354-363. [Crossref]
- Eraly SA, Bush KT, Sampogna RV, Bhatnagar V, Nigam SK (2004) The molecular pharmacology of organic anion transporters: from DNA to FDA? Mol Pharmacol 65: 479-487. [Crossref]
- Marchetti S, Mazzanti R, Beijnen JH, Schellens JH (2007) Concise review: Clinical relevance of drug drug and herb drug interactions mediated by the ABC transporter ABCB1 (MDR1, P-glycoprotein). Oncologist 12: 927-941. [Crossref]
- Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, et al. (2005) A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci USA 102: 17923-17928. [Crossref]
- Aleksunes LM, Cui Y, Klaassen CD (2008) Prominent expression of xenobiotic efflux transporters in mouse extraembryonic fetal membranes compared with placenta. Drug Metabol Dispos 36: 1960-1970. [Crossref]
- Pelkonen O, Mäenpää J, Taavitsainen P, Rautio A, Raunio H (1998) Inhibition and induction of human cytochrome P450 (CYP) enzymes. Xenobiotica 28: 1203-1253. [Crossref]
- Gonzalez FJ, Kasper CB (1982) Cloning of DNA complementary to rat liver NADPH-cytochrome c (P-450) oxidoreductase and cytochrome P-450b mRNAs. Evidence that phenobarbital augments transcription of specific genes. J Biol Chem 257: 5962-5968. [Crossref]
- Adesnik M, Bar-Nun S, Maschio F, Zunich M, Lippman A, et al. (1981) Mechanism of induction of cytochrome P-450 by phenobarbital. J Biol Chem 256: 10340-10345. [Crossref]
- Campana C, Regazzi MB, Buggia I, Molinaro M (1996) Clinically significant drug interactions with cyclosporin. An update. Clin Pharmacokinet 30: 141-179. [Crossref]
- Klotz U, Ammon E (1998) Clinical and toxicological consequences of the inductive potential of ethanol. Eur J Clin Pharmacol 54: 7-12. [Crossref]
- Wang H, LeCluyse EL (2003) Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet 42: 1331-1357. [Crossref]
- Xie W, Uppal H, Saini SP, Mu Y, Little JM, et al. (2004). Orphan nuclear receptor-mediated xenobiotic regulation in drug metabolism. Drug Discov Today 9: 442-449. [Crossref]
- Zhou G, Cummings R, Li Y, Mitra S, Wilkinson SA, et al. (1998). Nuclear receptors have distinct affinities for coactivators: characterization by fluorescence resonance energy transfer. Mol Endocrinol 12: 1594-1604. [Crossref]
- Blumberg B, Sabbagh W Jr, Juguilon H, Bolado J Jr, van Meter CM, et al. (1998) SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev 12: 3195-3205. [Crossref]
- Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, et al. (1998) Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A 95: 12208-12213. [Crossref]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, et al. (1998) The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest 102: 1016-1023. [Crossref]
- Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA (2001) Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol 60: 427-431. [Crossref]
- Gerbal-Chaloin S, Pascussi JM, Pichard-Garcia L, Daujat M, Waechter F, et al. (2001) Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab Dispos 29: 242-251. [Crossref]
- Moore JT, Kliewer SA (2000) Use of the nuclear receptor PXR to predict drug interactions. Toxicology 153: 1-10. [Crossref]
- Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, et al. (2002) Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol 16: 977-986. [Crossref]
- Ekins S, Erickson JA (2002) A pharmacophore for human pregnane X receptor ligands. Drug Metab Dispos 30: 96-99. [Crossref]
- Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, et al. (2000) St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci U S A 97: 7500-7502. [Crossref]
- Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, et al. (2000) Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 406: 435-439. [Crossref]
- Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, et al. (1994) A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol 14: 1544-1552. [Crossref]
- Forman BM, Tzameli I, Choi HS, Chen J, Simha D, et al. (1998) Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature 395: 612-615. [Crossref]
- Honkakoski P, Zelko I, Sueyoshi T, Negishi N (1998). The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol 18: 5652-5658. [Crossref]
- Gerbal-Chaloin S, Daujat M, Pascussi JM, Pichard-Garcia L, Vilarem MJ, et al. (2002) Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J Biol Chem 277: 209-217. [Crossref]
- Goodwin B, Hodgson E, D'Costa DJ, Robertson GR, Liddle C (2002) Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol Pharmacol 62: 359-365. [Crossref]
- Honkakoski P, Negishi M (1998) Protein serine/threonine phosphatase inhibitors suppress phenobarbital-induced Cyp2b10 gene transcription in mouse primary hepatocytes. Biochem J 330: 889-895. [Crossref]
- Pascussi JM, Gerbal-Chaloin S, Fabre JM, et al. (2000) Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: consequences on cytochrome P450 gene regulation. Mol Pharmacol 58: 1441-1450. [Crossref]
- Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, et al. (1999) Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol 19: 6318-6322. [Crossref]
- Sueyoshi T, Negishi M (2001) Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol 41: 123-143. [Crossref]
- Moore LB, Parks DJ, Jones SA, et al. (2000) Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem 275: 15122-15127. [Crossref]
- Tzameli I, Pissios P, Schuetz EG, Moore DD (2000) The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol 20: 2951-2958. [Crossref]
- Smirlis D, Muangmoonchai R, Edwards M, Phillips IR, Shephard EA (2001) Orphan receptor promiscuity in the induction of cytochromes p450 by xenobiotics. J Biol Chem 276: 12822-12826. [Crossref]
- Benedict WF, Considine N, Nebert DW (1973) Genetic differences in aryl hydrocarbon hydroxylase induction and benzo(a)pyrene-produced tumorigenesis in the mouse. Mol Pharmacol 9: 266-277. [Crossref]
- Poland A, Glover E, Kende AS (1976) Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem 251: 4936-4946. [Crossref]
- Hankinson O (1995) The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 35: 307-340. [Crossref]
- Burbach KM, Poland A, Bradfield CA (1992) Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A 89: 8185-8189. [Crossref]
- Nebert DW, Jones JE (1989) Regulation of the mammalian cytochrome P1-450 (CYP1A1) gene. Int J Biochem 21: 243-252. [Crossref]
- Diaz D, Fabre I, Daujat M, Saint Aubert B, Bories P, et al. (1990) Omeprazole is an aryl hydrocarbon-like inducer of human hepatic cytochrome P450. Gastroenterology 99: 737-747. [Crossref]
- Fuhr U (2000) Induction of drug metabolising enzymes: pharmacokinetic and toxicological consequences in humans. Clin Pharmacokinet 38: 493-504. [Crossref]
- Issemann I, Green S (1990) Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347: 645-650. [Crossref]
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, et al. (1998) Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98: 2088-2093. [Crossref]
- Forman BM, Chen J, Evans RM. (1997). Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A 94: 4312-4317. [Crossref]
- Schmiedlin-Ren P, Thummel KE, Fisher JM, et al. (1997) Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol Pharmacol 51: 741-754. [Crossref]
- Drocourt L, Ourlin JC, Pascussi JM, Maurel P, Vilarem MJ (2002) Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J Biol Chem 277: 25125-25132. [Crossref]
- Chen KS, DeLuca HF (1995) Cloning of the human 1 alpha,25-dihydroxyvitamin D-3 24-hydroxylase gene promoter and identification of two vitamin D-responsive elements. Biochim Biophys Acta 1263: 1-9. [Crossref]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, et al. (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6: 517-526. [Crossref]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383: 728-731. [Crossref]
- Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, et al. (1995) Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81: 687-693. [Crossref]
- Cheung C, Akiyama TE, Kudo G, Gonzalez FJ (2003) Hepatic expression of cytochrome P450s in hepatocyte nuclear factor 1-alpha (HNF1alpha)-deficient mice. Biochem Pharmacol 66: 2011-2020. [Crossref]
- Jover R, Bort R, Gomez-Lechon MJ, Castell JV (2001) Cytochrome P450 regulation by hepatocyte nuclear factor 4 in human hepatocytes: a study using adenovirus-mediated antisense targeting. Hepatology 33: 668-675. [Crossref]
- Corchero J, Granvil CP, Akiyama TE, Hayhurst GP, Pimprale S, et al. (2001) The CYP2D6 humanized mouse: effect of the human CYP2D6 transgene and HNF4alpha on the disposition of debrisoquine in the mouse. Mol Pharmacol 60: 1260-1267. [Crossref]
- Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, et al. (2003) The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 9: 220-224. [Crossref]
- Bort R, Gómez-Lechón MJ, Castell JV, Jover R (2004) Role of hepatocyte nuclear factor 3 gamma in the expression of human CYP2C genes. Arch Biochem Biophys 426: 63-72. [Crossref]
- Jover R, Bort R, Gómez-Lechón MJ, Castell JV (1998) Re-expression of C/EBP alpha induces CYP2B6, CYP2C9 and CYP2D6 genes in HepG2 cells. FEBS Lett 431: 227-230. [Crossref]
- Xu C, Li CY, Kong AN (2005) Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res 28: 249-268. [Crossref]
- Gardner-Stephen D, Heydel JM, Goyal A, Lu Y, Xie W, et al. (2004) Human PXR variants and their differential effects on the regulation of human UDP-glucuronosyltransferase gene expression. Drug Metab Dispos 32: 340-347. [Crossref]
- Staud F, Cerveny L, Ahmadimoghaddam D, Ceckova M (2013) Multidrug and toxin extrusion proteins (MATE/SLC47); role in pharmacokinetics. Int J Biochem Cell Biol 45: 2007-2011. [Crossref]