Abstract
The onset of spontaneous seizures in temporal lobe epilepsy is associated with an altered hippocampal circuitry. Neurochemical investigations have demonstrated that changes also occur in corticostriatal synaptic pathways. Since the striatum plays a major role in procedural learning, it is of particular interest whether dyskinetic motor dysfunction sometimes associated with epilepsy is indeed due to epilepsy-associated changes of basal ganglia function. Here, we analyzed corticostriatal long-term potentiation (LTP) in brain slices from pilocarpine-treated rats 4-10 weeks after status epilepticus. We stimulated corticostriatal fibers and recorded field potentials in the dorsomedial striatum. We found that LTP was significantly enhanced in slices from pilocarpine-treated rats as compared to control preparations. In chronically epileptic tissue, GABAA receptor blockade was not able to significantly enhance LTP any further. By contrast, in control slices it was able to strongly boost LTP. We conclude that epilepsy has a profound effect on the corticostriatal synaptic pathway, and that GABAA receptors lose their regulatory and suppressive role in synaptic plasticity in the dorsomedial striatum.
Key words
corticostriatal LTP, gabazine, excitatory postsynaptic, potential, pilocarpine
Introduction
Understanding the pathogenesis of temporal lobe epilepsy largely relies on the use of animal models such as the experimental epilepsy which occurs after a pilocarpine- induced status epilepticus [1]. Virtually all studies using animal models of epilepsy have focused on the hippocampus which is primarily affected [2,3]. Striatal pathology in experimental epilepsy, in turn, has generally not received the same level of attention. Only a few reports have addressed this issue and provided evidence that changes do occur within the basal ganglia. Activity-dependent changes in synaptic strength such as long-term potentiation (LTP) are widely accepted as key mechanisms for information storage in the brain [4], and LTP can indeed be induced at corticostriatal glutamatergic synapses [5,6]. The striatum is a brain region that is critical for controlling e.g. voluntary motor behaviors, formation of habits, drug addiction [7-9], and the dorsomedial part of the striatum has been shown to be key area for dystonic pathology in experimental models [10]. Therefore, synaptic plasticity of dorsomedial corticostriatal synapses appears to represent an adequate neural correlate of motor learning processes [5], and a possible marker of dystonic pathology [10]. Recently, we have reported that LTP in the dorsomedial striatum was profoundly upregulated in chronically epileptic tissue [11]. In the present study, we asked whether GABAA receptors might also be involved in alterations of synaptic plasticity following pilocarpine-induced status epilepticus. We confirmed an enhancement of LTP in epileptic tissue, and more importantly found this enhancement could be mimicked by GABAA receptor blockade in control slices.
Experimental procedure
Status epilepticus model
A sustained status epilepticus (SE) was induced in young male Wistar rats (~30 days, 140-150 g; Charles River, Sulzfeld, Germany). First, all animals received methylscopolamine nitrate (1 mg/kg, i.p.) to reduce peripheral cholinergic effects. After 30 minutes, pilocarpine hydrochloride was applied (340 mg/kg, i.p., in 0.9% saline) to induce SE which was terminated 40 minutes after onset by injection of diazepam (4-10 mg/kg, i.p.). If SE did not develop within 60 minutes, animals were reinjected (170 mg/kg, i.p.). Animals of the control group were identically treated, but were injected with saline instead of pilocarpine. Rats were tended and fed with glucose solution for 1 day and kept in separate cages. All procedures were performed according to national and international guidelines on the ethical use of animals (European Council Directive 86/609/EEC).
Brain slice preparation
Electrophysiological experiments were conducted 4-10 weeks after SE. Preparation of horizontal rat brain slices followed procedures as described earlier [12]. Briefly, the brains were quickly removed and chilled in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM) NaCl 125, NaHCO3 26, KCl 4, NaH2PO4 1.25, CaCl2 2, MgCl2 1.3 and glucose 10 gassed with carbogen (95% O2, 5% CO2). The brain was trimmed on the dorsal side at an angle of approximately 40° from the horizontal and glued to a vibratome based on that side (Integraslice 7550MM, Campden Instruments Ltd). From this slanted tissue block, angulated 500 μm slices were made which contained the motor cortex and entire striatum, with connections between these two regions still intact. Immediately after the slicing, slices were transferred to the interface-type recording chamber and were incubated at room temperature for at least 1 hour and then for another 1 hour at 32-33°C. The remaining slices were transferred to an incubation submersion-type bath filled with ACSF.
Electrophysiological recordings
Recordings were performed following an equilibration of at least 2.5-3 hours. Field excitatory postsynaptic potentials (fEPSPs) were obtained from the dorsomedial striatum using a glass micropipette recording electrode filled with ACSF. Signals were processed and digitized using a micro1401 A/D converter controlled by Signal 2.16 (Cambridge Electronic Design, Cambridge, UK). The white matter of the cortex was stimulated at a rate of 0.033 Hz with a bipolar insulated platinum wire electrode (50 μm diameter). Stimulus strength was set at 50% of saturating intensity. Following 10-15 minutes of stable baseline, long-term potentiation (LTP) was induced with high- frequency stimulation (HFS, 4 × 100 Hz, 1 s duration, with 10 s intervals) at the same stimulation intensity as used for single fEPSP recordings. The initial slopes of the fEPSPs were measured, and then expressed as a percentage of baseline level, calculated from an average of the last 10 minutes of the baseline recording period. The degree of LTP for each experiment was measured as the average of the last 10 minutes of the post-HFS, i.e. from data 50 to 60 minutes after HFS. Data were expressed as means ± SEM. Statistical analysis was done with the SigmaStat software (SPSS; Chicago, IL, USA), and differences were considered significant when P<0.05. All chemicals used for solutions were purchased from Sigma (Taufkirchen, Germany). The GABAA receptor blocker gabazine was purchased from Tocris (Bristol, UK). A stock solution of 100 mM gabazine was prepared in bi-distilled water. For the experiments, a final concentration of 3 µM gabazine was used. We chose this concentration, since we aimed to stay well below 10 µM to spare tonic GABAergic activity [13], and to get closer to the ID50 of 0.44 µM reported for cultured, isolated neurones [14], which, however, would necessitate unrealistically prolonged incubation times in 500 µm thick slice preparations.
Results
Corticostriatal axons terminate on both medium spiny neurons and GABAergic, fast- spiking striatal interneurons [15]. Hence, GABAergic activity within the striatum might influence the propensity to obtain corticostriatal LTP. Since we were interested in the contribution of GABAA receptor activation to corticostriatal LTP in control and epileptic tissue, we first confirmed our previous results, i.e. the LTP enhancement in slices taken from pilocarpine-treated rats. When high-frequency stimulation (HFS) was applied to control slices, we found an increase in the fEPSP slope to 110 ± 6% of baseline after 60 minutes of follow-up (n=12, open symbols in Figures 1A and 1D). As has been reported previously [11], slices from pilocarpine-treated animals showed significantly enhanced levels of LTP (156 ± 10% of baseline, n=9, closed symbols in Figures 1A and 1D; P<0.01). This difference, however, was completely lost when the specific GABAA receptor blocker gabazine (3 µM) was used throughout the experiment. In gabazine-preincubated slices, the time course of the fEPSP potentiation following HFS was similar in both experimental groups. After 1 hour of follow-up, LTP levels reached 162 ± 13% of baseline (n=14) in controls and 163 ± 8% of baseline (n=12) in pilocarpine-treated rats (Figures 1B and 1D). Interestingly, these values were almost identical with the LTP level obtained in epileptic tissue without gabazine. Thus, GABAA receptor-mediated inhibition was present under control conditions, substantially controlling and suppressing LTP induction. In the striatum of chronically epileptic animals, however, this GABAergic inhibition appeared to be down-regulated since gabazine was no longer able to enhance LTP levels. One potential pitfall in interpreting these results is that LTP was determined relative to baseline levels which themselves were established after gabazine-preincubation. We therefore repeated these experiments and applied gabazine during the recording to control for pre-incubation effects. Also in these experiments, GABAergic control of LTP is evident only in control slices: Following 25 minutes of GABAA receptor inhibition, fEPSP slopes increased to 126 ± 12% of baseline in control tissue (n=6, Figures 1C and 1D), and to 125 ± 23% of baseline in slices from pilocarpine-treated rats (n=6). These data suggest that tonic GABAergic inhibition is preserved in both control tissue and the epileptic striatum. More importantly, however, the potentiation of fEPSP slopes after HFS again reached similar values in both experimental groups (control: 197 ± 36% of baseline, n=6; pilocarpine: 198 ± 29% of baseline, n=6; Figures 1C and 1D), as in pre-incubated slices. Thus, phasic inhibition occurring during the HFS paradigm rather than tonic inhibition might be functionally down-regulated in the epileptic striatal network.
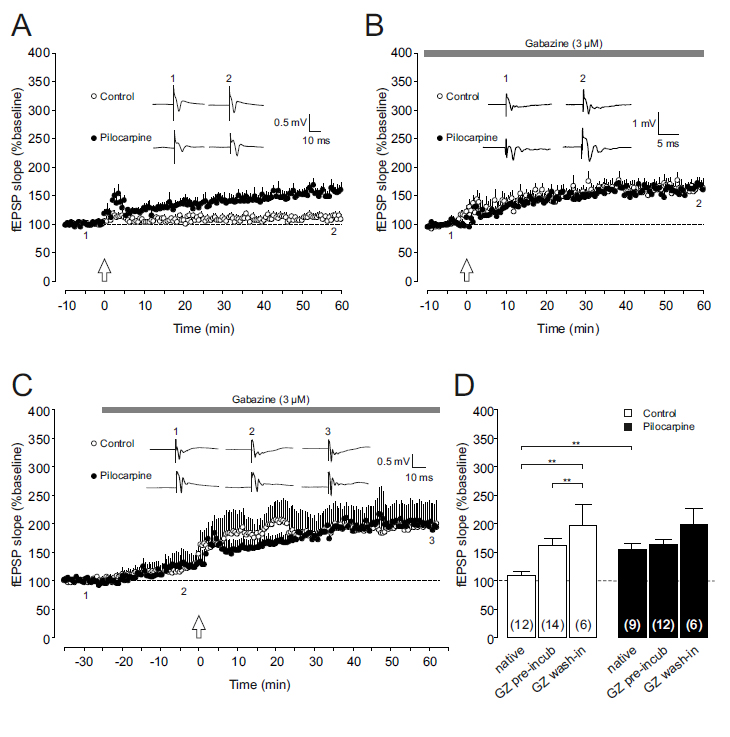
Figure1.Effect of GABAAreceptor inhibition on corticostriatalLTP.
(A)Long-termpotentiation(LTP)atcorticostriatalsynapsesfromchronicallyepileptic rats(Pilocarpine,closedcircles)issignificantlyenhancedcomparedtocontroltissue (Control,opencircles).Dotplots(means±SEM)ofrelativefieldpotential(fEPSP) slopesofcorticalafferentstothedorsomedialstriatumareshownfrom10minutes beforeto60minutesafterhigh-frequencystimulation(HFS,indicatedbyanarrow). Insets:Originalfieldpotentialrecordingsfromdorsomedialstriatumobtainedattime points1and2duringthecourseoftheexperiment(i.e.shortlybefore and~60 minutesafterHFS).
(B,C)LTPisenhancedincontrolpreparations(opencircles) underconditionsofGABAAreceptorblockade,butisunalteredinslicesfrom pilocarpine-treatedrats(closedcircles).Dotplots(means±SEM)ofrelativefEPSP slopesareshownforgabazine-preincubatedslices(panelB)aswellasfor experimentsdocumentingthe temporal developmentofgabazine treatmentto assess fEPSPchangesinducedbyGABAAreceptorinhibition(panelC).HFSwasappliedat timepoint“0”(indicatedbyanarrow).Sampletracesdemonstratetheincreaseofthe fEPSPslopeinthepresenceofgabazine(3µM)inbothcontrolandpilocarpine tissue(panelC)aswellasafterhigh-frequencystimulation(panelsBandC).
(D)Bar graph summarizingtheLTPvalues(i.e.thefEPSPslopesat~60minafterHFS, relativetobaseline)fornativeconditions,gabazine-preincubatedslicesandslices withdevelopinggabazineeffectincontroltissue(openbars)andtissuefrom chronicallyepileptic animals (closedbars).
Discussion
During the process of epileptogenesis, a number of alterations have been reported in hippocampal and parahippocampal brain areas [16]. However, extratemporal neural structures such as the basal ganglia are increasingly recognized as being also involved in chronic synaptic changes that occur during epileptogenesis. In our previous work, we addressed corticostriatal LTP in pilocarpine-treated rats and observed that LTP was significantly enhanced in the striatum of chronically epileptic rats, while it was hardly expressed in normal tissue [11]. The present study confirmed these results and further, expands on the role of GABAA receptor mediated transmission in corticostriatal LTP. The major finding of the present study is that gabazine enhanced LTP in control tissue, whereas in pilocarpine-treated rats LTP was practically unaltered. At first sight, this might suggest disinhibition in control tissue leading to further increases in excitability (and hence LTP), while in epileptic tissue, a depolarizing GABAergic component of synaptic transmission emerging under pathological conditions – as speculated e.g. in striatal neurons in a model of absence epilepsy [17] – might be involved. However, two findings argue against this: i. in the cited absence model, phasic GABAergic activity during spike-and-wave discharges led to shunting inhibition nevertheless, and not to augmentation of responses [17], and ii. in the present study, GABAergic receptor suppression led to significant increases in fEPSP slopes in both epileptic and control tissue, suggesting substantial tonic inhibition being preserved also under the condition of chronic hyperexcitability (e.g. epilepsy). One can speculate that this tonic inhibition is emerging as a protective response to the pathological discharges, and that once it is lifted (by receptor blockade), it limits the dynamic range of plasticity, since the system is already close to saturation. By contrast, in healthy tissue, phasic inhibition appears to prevail over tonic one, and is indeed limiting LTP induction (likely via coactivation of cortical projections of GABAergic striatal neurons known to exert strong feedforward inhibition [17,18]. Here, the strong modulation of LTP by GABAA receptor blockade might suggest that LTP is at least in part resulting from the activity-dependent modulation of intrastriatal inhibitory networks [19-21].
In conclusion, our results indicate that epileptogenesis may also involve brain areas not directly associated with the origin of epileptic seizures, and more specifically, that phasic inhibitory control keeps LTP at bay under normal conditions, and is lost in chronically epileptic tissue.
Acknowledgements
The authors thank Katrin Porath, Tina Sellmann, Ulrike Mikkat and Hanka Schmidt for their excellent technical assistance and animal care. This study was supported by the German Research Council (DFG, grant KO1179/4-2) and the Federal Ministry for Education and Science (BMBF, grant 01GQ0751).
References
2021 Copyright OAT. All rights reserv
- Turski L, Cavalheiro EA, Bortolotto ZA, Ikonomidou-Turski C, Kleinrok Z, et al. (1988) Dopamine-sensitive anticonvulsant site in the rat striatum.J Neurosci8: 4027-4037. [Crossref]
- Curia G, Longo D, Biagini G, Jones RS, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy.J Neurosci Methods 172: 143-157. [Crossref]
- Scorza FA, Arida RM, Naffah-MazzacorattiMda G, Scerni DA, Calderazzo L, et al. (2009) Thepilocarpine model of epilepsy: what have we learned?An Acad Bras Cienc81: 345-365. [Crossref]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus.Nature361: 31-39. [Crossref]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G (1992) Long-term synaptic depression in the striatum: physiological and pharmacological characterization.J Neurosci12: 4224-4233. [Crossref]
- Partridge JG, Tang KC, Lovinger DM (2000) Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum.J Neurophysiol84: 1422-1429. [Crossref]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M (1994) The basal ganglia and adaptive motor control.Science265: 1826-1831. [Crossref]
- Berke JD, Hyman SE (2000) Addiction, dopamine, and the molecular mechanisms of memory.Neuron25: 515-532. [Crossref]
- Yin HH, Knowlton BJ, Balleine BW (2004) Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning.Eur J Neurosci19: 181-189. [Crossref]
- Nobrega JN, Raymond R, Barlow K, Hamann M, Richter A (2002) Changes in AMPA receptor binding in an animal model of inborn paroxysmal dystonia.ExpNeurol176: 371-376. [Crossref]
- Avchalumov Y, Kirschstein T, Köhling R (2011) Altered physiology and pharmacology in the corticostriatal system in a model of temporal lobe epilepsy.Epilepsia52: 151-157. [Crossref]
- Köhling R, Koch UR, Hamann M, Richter A (2004) Increased excitability in cortico-striatal synaptic pathway in a model of paroxysmal dystonia.Neurobiol Dis16: 236-245. [Crossref]
- Yeung JY, Canning KJ, Zhu G, Pennefather P, MacDonald JF, et al. (2003) Tonically activated GABAA receptors in hippocampal neurons are high-affinity, low-conductance sensors for extracellular GABA.MolPharmacol 63: 2-8. [Crossref]
- Uchida I, Cestari IN, Yang J (1996) The differential antagonism by bicuculline and SR95531 of pentobarbitone-induced currents in cultured hippocampal neurons.Eur J Pharmacol307: 89-96. [Crossref]
- Wilson CJ (2007) GABAergic inhibition in the neostriatum.Prog Brain Res160: 91-110. [Crossref]
- Scharfman HE (2000) Epileptogenesis in the parahippocampal region. Parallels with the dentate gyrus.Ann N Y AcadSci 911: 305-327. [Crossref]
- Slaght SJ, Paz T, Chavez M, Deniau JM, Mahon S, et al. (2004) On the activity of the corticostriatal networks during spike-and-wave discharges in a genetic model of absence epilepsy.J Neurosci24: 6816-6825. [Crossref]
- Plenz D, Kitai ST (1998) Up and down states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex-striatum-substantianigraorganotypic cultures. J Neurosci18: 266-283. [Crossref]
- Caillard O, Ben-Ari Y, Gaiarsa JL (1999) Long-term potentiation of GABAergic synaptic transmission in neonatal rat hippocampus.J Physiol 518: 109-119. [Crossref]
- Gaiarsa JL, Caillard O, Ben-Ari Y (2002) Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance.Trends Neurosci25: 564-570. [Crossref]
- Pelletier JG, Lacaille JC (2008) Long-term synaptic plasticity in hippocampal feedback inhibitory networks.Prog Brain Res169: 241-250. [Crossref]