Paediatric mixed connective tissue disease (MCTD) is a rare and heterogeneous group of rheumatic disorders in children and young people, characterised by overlapping features of juvenile idiopathic arthritis (JIA), juvenile systemic lupus erythematosus (JSLE), systemic sclerosis (SSc), and juvenile dermatomyositis/polymyositis (JDM/PM) in the presence of high titres of anti-U1 ribonucleoprotein antibodies (RNP) [1,2]. Timely diagnosis and management of this condition has significant challenges posed by:
- The rarity of the condition
- Heterogeneity of presentation
- Lack of consensus regarding classification criteria
- Often long drawn out natural history where the disease evolves over a period of time
- Paucity of data in paediatric population
- Big variation in the response to treatment – excellent response of some of the common symptoms as against poor response of some of the others like sclerodactyly, oesophageal dysmotility, pulmonary artery hypertension and interstitial lung disease.
Many of these can be amply illustrated by the patients we are discussing in this article, who all were diagnosed with MCTD at our tertiary Paediatric Rheumatology centre in the last five years:
14-year-old girl, previously fit and well except for mild Raynaud’s phenomenon (RP), was initially referred with swelling and stiffness of fingers, which seemed to have settled following regular Naproxen by the time of clinic appointment. Her hands were felt to be normal on examination and ultrasound scan, blood tests including inflammatory markers and autoantibodies were all normal ; but she was kept under follow up. Swelling of fingers recurred seven months later, with no associated systemic symptoms. Examination revealed thickening and “bogginess” around proximal interphalangeal (PIP) and metacarpophalangeal (MCP) joints bilaterally. MRI scan showed moderate tenosynovitis affecting the flexor and extensor tendon sheaths with moderate fluid around all MCP and PIP joints (Figure 1). Her blood tests were unremarkable again. She was diagnosed as Rheumatoid Factor (RF) negative polyarticular Juvenile Idiopathic Arthritis (JIA).
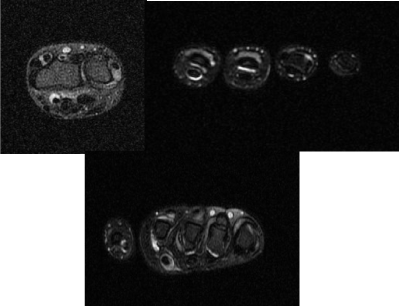
Figure 1. MRI scan of hands showing tenosynovitis affecting flexor and extensor tendon sheaths and effusion around MCP and PIP joints.
Following a pulse of intravenous Methyl Prednisolone (IVMP) at 30mg/kg for 3 days, she was commenced on a weaning course of oral prednisolone and weekly subcutaneous Methotrexate (MTX) injections (15mg), with very good result and complete resolution of symptoms.
However, eight months later she developed erythematous, confluent, vasculitic rash on soles of her feet (Figure 2), while still on Methotrexate.
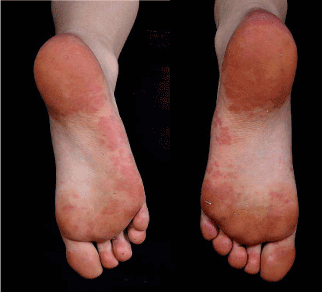
Figure 2. Erythematous, confluent vasculitic rash over the soles of the feet.
When reviewed urgently in our combined dermatology clinic, a diagnosis of chilblain lupus was considered. Investigations revealed normal FBC and ESR, mildly elevated muscle enzymes [Creatinine kinase (CK) 800 U/L], low complements C3 and C4, positive lupus anticoagulant [dilute Russell's viper venom time (dRVVT)] and anti-double stranded DNA (ds DNA) antibodies; and strongly positive Anti-Nuclear Antibodies (ANA) with fine, speckled pattern, IgG cardiolipin and anti-Ribonucleoprotein (RNP) antibodies. At this point her diagnosis was revised as MCTD with predominant lupus. Her lung function and echocardiogram were normal with no proteinuria. She received a further pulse of IVMP; the weekly dose of MTX was increased to 20mg and was commenced on Hydroxychloroquine (HCQ) and Aspirin.
After initial response (probably to steroids), she developed the classical butterfly malar rash and some erythematous vasculitic lesions over her ears, arms and palms (Figure 3). She was still systemically well doing about 7 hours of swimming every week. Her C4 had normalised; but continued to have low C3 and positive autoantibodies, as before. She was given another pulse of IVMP and was commenced on Mycophenolate Mofetil (MMF), in addition to existing medications, with good response and is currently in remission.
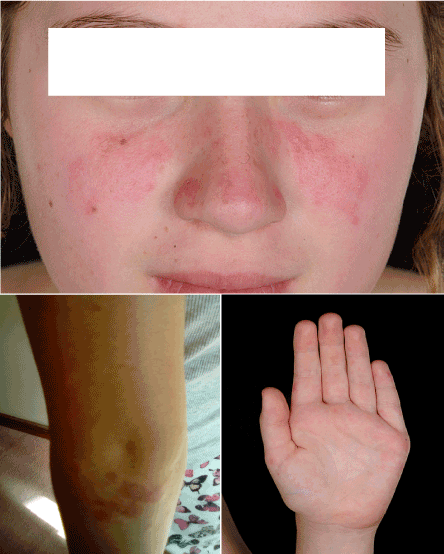
Figure 3. Classical butterfly rash and erythematous vasculitic lesions over her arms and palms.
A 16-year-old girl presented with pain and stiffness in multiple joints along with swollen hands for four weeks. She reported extreme fatigue and exhaustion, recent onset of mouth ulcers, worsening RP and menorrhagia, with no rashes. Her fingers were diffusely swollen and restricted with tight, shiny skin, along with swelling and restriction of shoulders, hips, ankles and subtalar joints. A diagnosis of a CTD was considered and investigations revealed high ESR, muscle enzymes, positive autoantibodies – RF [but negative anti cyclic citrullinated peptide antibodies(CCP)], ANA (fine, speckled pattern), ds DNA and anti RNP antibodies. She had a multi-system work up including Chest X ray, electrocardiogram (ECG), echocardiogram, pulmonary function and urine Protein Creatinine Ratio (PCR), all of which were normal. The diagnosis of MCTD was made, in view of features of myositis, lupus and sclerodactyly. She was given two pulses of IVMP followed by weaning dose of oral Prednisolone, HCQ, Aspirin and weekly subcutaneous MTX. She responded well to treatment and is in remission currently.
9-year-old girl was referred with recurrent episodes of fever, sore throat, lymphadenopathy, rashes, joint pain and fatigue, each lasting 2-3 weeks. She was previously fit and well, except having had multiple surgeries for developmental dysplasia (DDH) of right hip. These symptoms were first noticed when she was recovering from surgery, as an erythematous rash over the surgical wound which subsequently spread to the torso, associated with pyrexia, which did not respond to antibiotics. These symptoms were thought to be secondary to antibiotics and she slowly recovered once they were stopped, although was very tired on return to school. She developed severe RP that winter, followed by fever, sore throat and joint pain, with mild anaemia and high ESR; and a diagnosis of glandular fever was suspected. Pain in knees, ankles and hands persisted, along with extreme fatigue, which prompted the referral. On examination, she had a faint malar rash, puffy and restricted fingers (could not make a fist) with shiny skin (Figure 4); rest of the examination including muscle strength was unremarkable.
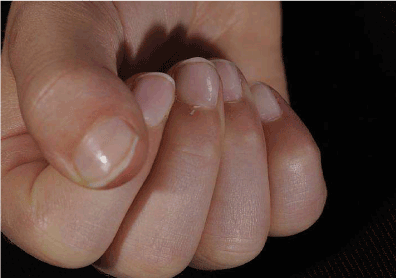
Figure 4. Puffy, restricted, shiny fingers – unable to make a fist.
She had elevated ESR of 34mm/hr, with normal FBC and complement levels. Autoantibody screen revealed strongly positive RF (309.7 kIU/L), ANA with coarse, speckled pattern and anti RNP antibodies; ds DNA was negative. An extended infection screen was negative on serology. She had three monthly pulses of IVMP, along with weekly subcutaneous MTX injections and HCQ, with a diagnosis of RF positive polyarticular JIA possibly overlapping with evolving CTD. She went into remission; but the joints flared while she was switched to oral MTX due to intolerance. She was commenced on Etanercept injections, alongside HCQ. She stayed in remission for nearly two years before she started experiencing recurrent episodes of sore throat, swollen glands and fatigue, lasting 2-3 weeks with poor response to antibiotics. During one of those episodes, she was noted to have swollen parotid glands; not associated with rashes or joint symptoms. Her ESR was elevated at 28mm/hr; with positive RF (89 kIU/L), ANA and anti RNP antibodies; but negative anti-CCP, ds DNA, anti Ro and anti La antibodies. Schirmer's test was positive (moderately dry eyes) while ultrasound scan (USS) of parotid glands showed numerous abnormal hypoechoic circular areas (Figure 5) suggestive of Sjogren's. With a diagnosis of MCTD with secondary Sjogren's syndrome, MMF was commenced, with poor response, following which she was switched to Rituxumab, to which she responded.
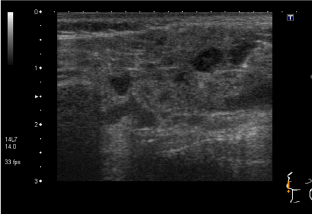
Figure 5a. Abnormal hypoechoic circular areas on USS of parotid glands
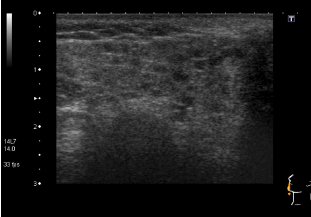
Figure 5b. Abnormal hypoechoic circular areas on USS of parotid glands.
A 16-year-old girl was referred with 2-year history of painful swelling of hands and ankles, cold hands and feet despite maximum thermal protection, and exhaustion affecting her school attendance. She also had muscle pain and weakness, constipation, and hair loss. Her father and paternal grandparents had rheumatoid arthritis (RA), while mother had idiopathic pulmonary fibrosis. Her examination revealed arthritis in ankles, wrists and right elbow in addition to swollen, restricted and cold fingers. She also had proximal muscle weakness with mild alopecia of scalp, but no rashes. Her blood tests showed raised ESR (98mm/hr) and muscle enzymes (CK 5825 U/L and LDH 1824 U/L) with normal FBC and CRP. She had strongly positive ANA with speckled pattern and positive anti RNP antibodies, while she was negative for RF, anti CCP and ds DNA antibodies. Her multi system work up showed normal echocardiogram, normal MRI scan of lower limb muscles (done after the first pulse of IV methyl prednisolone) and a restrictive pattern on lung function. Following a pulse of methyl prednisolone, she was started on weaning course of oral Prednisolone and weekly MTX injections (20mg) with good clinical and laboratory response. However she defaulted her review appointments, stopped the medications and presented with a flare few months later - struggling with activities of daily living, with swollen hands (no synovitis on USS), muscle weakness and erythematous skin rash on exposure to sun. She was given pulse of steroids again, and restarted on MTX injections along with daily HCQ tablets. Poor compliance and engagement continued before another flare, associated with worsening lung function (Figure 6), reduced transfer factors and early interstitial changes on High Resolution CT (HRCT) scan (Figure 7). Following multiple pulses of IV methyl prednisolone, she was commenced on slow weaning dose of oral Prednisolone and MMF added to HCQ, which induced clinical remission, although her muscle enzymes are still high.
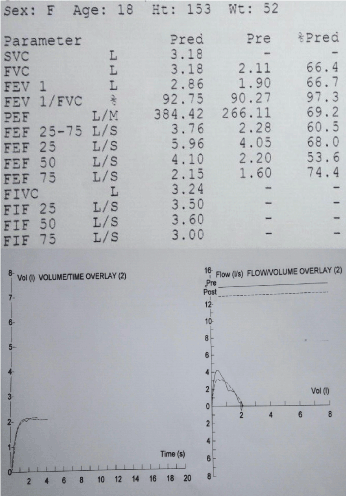
Figure 6. The spirometry demonstrating restrictive picture with Forced Vital Capacity (FVC) 66.4% of expected.
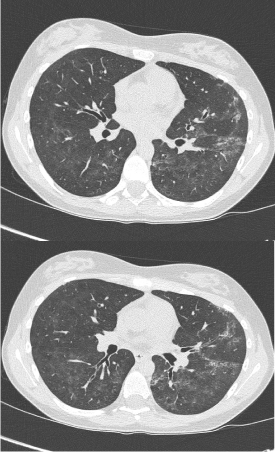
Figure 7. HRCT scan showing multiple focal areas of coarse interstitial change
A 11-year-old boy presented with widespread joint pain and stiffness for a year and worsening RP. Examination revealed florid polyarthritis affecting over 25 joints, both small and large, with no other systemic features except RP. He had strongly positive ANA and anti RNP antibodies; but FBC, inflammatory markers, complement, and rest of the autoantibody profile were normal. He had normal echocardiogram, spirometry and urine protein: creatinine ratio. With a diagnosis of polyarticular JIA with evolving CTD, he was started on weekly Methotrexate injections, after a pulse of IV methyl prednisolone followed by tapering course of oral Prednisolone. After initial response, his joints flared after 6 months, when he also developed livedo reticularis, following which he received intra articular steroid injections, the dose of MTX was increased and HCQ added. Lupus anticoagulant, anti-cardiolipin antibodies and thrombophilia screen were negative. He further developed well demarcated, blistering, erythematous eruptions on the thighs and arms, confirmed to be erythema Multiforme on biopsy. The skin lesions settled down after a short course of oral steroids. After a period of stability of joints and skin, he was switched to oral MTX, before stopping it a year later, as joints remained in remission .He still has troublesome RP and continues on HCQ. His multi system screen continues to be normal with an unchanged autoantibody profile.
A 9-year-old girl with long standing history of intermittent pain in her joints was referred by the paediatric team due to positive ANA and anti RNP antibodies, following investigation of an episode of erythema multiforme (presumed to be streptococcal). The pain mainly affected her ankles, elbows and fingers, with transient, intermittent swelling and stiffness in the morning. She reported worsening fatigue of recent onset and RP; but no rashes, mouth ulcers or any systemic symptoms. Her examination was normal (including skin, joints and muscles) except for mild RP. Her blood tests revealed strongly positive ANA and anti RNP antibodies with marginally elevated muscle enzymes and IgG but normal FBC, inflammatory markers, complement and urine PCR. CXR, echocardiogram and lung function were normal too. She then developed an intermittent photosensitive rash over her arms and legs, diagnosed as urticarial vasculitis following biopsy. She was commenced on HCQ at this stage with good response. She then developed a faint but persistent erythematous malar rash sparing her nasolabial folds, along with an odd mouth ulcer, although her blood tests remained normal, except for the autoantibody profile. After being relatively stable on HCQ for three years, she reported worsening RP, fatigue and exercise tolerance with increasing frequency of rashes and mouth ulcers. The faint malar rash was unchanged; but her joints and muscles were normal. Her blood tests revealed marginal increase in CK, slowly dropping lymphocyte count and persistently elevated ANA and anti RNP antibodies;C3, C4, inflammatory markers, lung function and echocardiogram were normal. In view of the worsening symptoms, she was recently commenced on Azathioprine with good response.
Table 1 summarises the challenges in the diagnosis of MCTD in children (median time from onset 16 months), due to a mixture of factors mentioned earlier. Only cases 2 & 4 sounded out the diagnosis at the outset; the rest were managed as JIA or lupus before the clinical and autoimmune profile changed over a variable period of time. The clinical profile of Case 6, even after five years since the presentation, is still evolving and does not yet satisfy the diagnostic criteria. RP and synovitis were the most frequent features, followed by fatigue, not dissimilar to literature [1,3]. The myositis (present in 50%) was mild, except in one; but none had symptoms to suggest oesophageal dysmotility, although they all received Proton Pump Inhibitors (PPI) in the initial phase of treatment. Case 3 had Secondary Sjogren’s Syndrome (seen in one third) [4] and 4 had interstitial lung disease (seen in about one third) [5]. None of them had renal involvement, which is reported less often in MCTD when compared to JSLE [2]. All but one had skin manifestations other than RP (malar rash only in 50%); it is interesting to note that 2/6 patients had erythema multiforme at some stage, which has been described in MCTD [6,7]. All the patients had very high titres of ANA (fine speckled pattern) and anti RNP antibodies; while one third had positive RF,ds DNA and hypocomplementaemia. All the patients responded to systemic corticosteroid therapy; but required a wide variety of agents to maintain remission – MTX (2/6), MMF (2/6), and Rituximab (1/6), in addition to HCQ.
Table 1. Summary of clinical profile of the 6 patients with MCTD
|
Case 1 |
Case 2 |
Case 3 |
Case 4 |
Case 5 |
Case 6 |
Total |
Raynauds |
+ |
++ |
++ |
++ |
++ |
++ |
6/6 (100%) |
Synovitis |
++ |
++ |
++ |
++ |
++ |
+ (transient) |
6/6 (100%) |
Swollen hands |
- |
++ |
++ |
++ |
- |
|
3/6 (50%) |
Myositis |
+ |
+ |
- |
++ |
- |
+ |
3/6 (50%) |
Malar rash |
++ |
- |
+ |
- |
- |
+ |
3/6 (50%) |
Other rashes |
Erythematous vasculitic rashes |
- |
Erythematous maculo-papular rash with fever with +ve Koebner |
Alopecia
Erythematous photosensitive skin rash |
Erythema multiforme
Livedo reticularis |
Erythema multiforme
Urticarial vasculitis
Mouth ulcers |
1/6 (17%) |
Lung involvement |
- |
- |
- |
++ |
|
|
|
Fatigue |
- |
++ |
++ |
++ |
+ |
++ |
5/6 (84%) |
Other clinical features |
|
|
Adenopathy +
Xerostomia
Xerophthalmia |
|
|
|
|
ANA |
++ fine speckled |
++ fine speckled |
++ fine speckled |
++fine speckled |
++ fine speckled |
++ fine speckled |
6/6 (100%) |
Anti RNP |
++ |
++ |
++ |
++ |
++ |
++ |
6/6 (100%) |
Anti-ds DNA |
++ |
++ |
- |
- |
- |
- |
2/6 (33%) |
Other autoantibodies |
Anti cardiolipin |
RF |
RF |
|
- |
|
|
Abnormal FBC |
- |
- |
- |
- |
- |
Lymphopaenia |
1/6 (17%) |
Low complement |
++ |
++ |
- |
- |
- |
- |
2/6 (33%) |
Treatment |
MTX + MMF + HCQ |
MTX + HCQ |
Rituximab + HCQ |
MMF + HCQ |
MTX + HCQ |
Azathioprine + HCQ |
|
Time to diagnosis from onset |
14 months |
1 month |
56 months |
24 months |
18 months |
12 months |
Median 16 months |
From a practical point of view, MCTD is suspected in patients with positive anti RNP antibodies in the presence of clinical features and/ or investigations suggestive of two of the following three – JSLE, JDM and scleroderma. We do a thorough multi-system screening in these patients – urine for PCR, Chest X-ray (CXR), basic lung function (FEV1 and FVC), Electrocardiogram (ECG) and Echocardiogram. These patients are reviewed regularly in our multi-disciplinary clinics, where specialist nurses take the lead on patient education and providing a helpline; and the therapists design and run the individualised rehabilitation programme to achieve the maximal physical fitness. We also work closely with colleagues in other paediatric subspecialties – nephrology, neurology, pulmonology, cardiology, radiology and dermatology – through meetings and/or combined clinics, to plan further investigations, as guided by the initial presentation and the baseline investigations, try and reach a consensus on therapeutic plan and follow up.
First described by Sharp et al. in 1972 [8], there was lot of debate whether MCTD was a distinct clinical entity, due to the overlap of symptoms with other rheumatological conditions. However, the emergence of distinct genetic (association with HLA DR4, which is distinct from those found most commonly in SLE, SSc, or PM/DM) and serological (high titres of anti U1 RNP antibodies in the absence of anti Sm antibodies, which are pathognomonic of JSLE) profiles [9] have helped to characterise MCTD better. None of the four classification criteria for MCTD published so far - Kasukawa’s, Alarcon-Segovia, Kahn and Sharp criteria – [10-13] have been validated for use in children. The studies comparing these [14,15] have provided conflicting results (Table 2).
Table 2. Classification Criteria for MCTD
|
Serological criteria |
Clinical criteria |
Diagnosis |
Alarcon-Segovia Diagnostic Criteria(1987) |
Positive anti U1 RNP at haemagglutination titer >1:1600 |
- Oedema of hands
- Synovitis
- Myositis
- Raynaud´s
- Acrosclerosis
|
Serological and at least 3 clinical features included synovitis or myositis |
Kusukawa Diagnostic Criteria(1987) |
Common Symptoms
Raynaud´s Phenomenon
Swollen fingers or hands
Presence of Anti U1 RNP |
Mixed findings
A. Systemic lupus erythematosus (SLE) like:
Polyarthritis
Pericarditis/pleuritis
Lymphadenopathy
Facial erythema
Leucopenia/thrombocytopenia
B. Scleroderma like:
Sclerodactyly
Pulmonary fibrosis
Esophageal dysmotility
C. Polymyositis like
Muscle weakness
High creatine phosphokinase (CPK)
Myopathic electromyogram (EMG) |
At least one common symptom, with positive U1 RNP antibodies and one or more findings in at least two of the three categories A, B, and C. |
Sharps Criteria (1987) |
Major Criteria
a1.Myositis, severe
2. Pulmonary involvement
a. CO diffusing capacity < 70% of normal
b. Pulmonary arterial hypertension
c. Proliferative vascular lesions in lung
biopsy
3. Raynaud’s phenomenon,
or esophageal hypomotility
4. Swollen hands observed or sclerordactyly
5. Highest observed anti-ENA≥1:10,000 and
anti U1 RNP positive and anti-Sm negative |
Minor Criteria
1. Alopecia
2. Leukopenia <4,000 WBC/mm3
3. Anemia ≤10.0 g/dL females, <12.0 g/dL males
4. Pleuritis
5. Pericarditis
6. Arthritis
7. Trigeminal neuropathy
8. Malar rash
9. Thrombocytopenia (<100,000/mm3
)
10. Myositis, mild
11. Swollen hands or history of swollen hand |
Definite 4 major criteria, and anti-U1 RNP positive, with anti-ENA ≥1:4000. Exclusion: anti-Sm
Probable 3 major criteria and anti-U1 RNP positive with anti ENA ≥1.1000
2 major criteria (include 1 or more from #1, #2, #3 and 2 minor criteria, and
anti-U1 RNP positive with anti- ENA ≥1.1000)
3 major criteria
Possible 2 major criteria and anti-U1 RNP positive with anti-ENA ≥1:100
1 major and 3 minor, and anti-U1 RNP positive with anti-ENA ≥1:100 |
Khan’s criteria |
Serological criteria:
Positive anti-RNP (U1 68kd) anti-RNP ≥ 1/2000) |
Clinical criteria:
1. Raynaud’s phenomenon (obligatory)
2. Synovitis
3. Myositis
4. Puffy hands |
Serological criteria
Plus ≥3 clinical criteria |
MCTD is one of the least common Paediatric Rheumatology conditions with frequency varying from 0.1% to 0.5% [1] in various studies. It is about six times commoner in girls with median age of onset of 11 years (range 4 – 16) [1,3].
The mechanism involves an environmental trigger causing an immune activation in a genetically predisposed individual (HLA-DR4 and HLA-B8). Both the innate and adaptive immune systems are involved in the development of MCTD [2] with 70-kD peptide of the Anti-U1RNP antigen playing a central role, reacting with both autoantibodies and T cells [1], and responsible for many of the core clinical features. Recent studies also implicate Vascular Endothelial Growth Factor (VEGF) [16] and Endogenous Toll Like Receptors (TLRs) [17] in the pathogenesis of MCTD.
Symptoms of MCTD evolve over a period of time that mean duration from first symptoms to the diagnosis is reported to be between 1.7 to 2 years [3,18]. RP and polyarthritis (usually non erosive) are the most common presenting symptoms, followed by other cutaneous manifestations (oedema of hands, JSLE or JDM rash) often with nailfold capillary changes, myositis (often subclinical or mild), oesophageal dysmotility and serositis (last two not observed in any of our patients) [1,2]. Renal involvement is less common than in JSLE; but more common in children with MCTD when compared to adults [5,19], as are haematological abnormalities. Although pulmonary function abnormalities are frequent even in asymptomatic patients, interstitial lung disease is reported only in about one third [19,20]. Pulmonary hypertension is less common in children with MCTD when compared to adults [1].
The serological hallmarks are high titres of Antinuclear antibodies and anti RNP antibodies (Anti-U1 RNP antibodies are more specific for MCTD); followed by RF (68-70%) and anti-ds DNA (20-40%) [2,3]. A number of other autoantibodies like Anti-Sm, anti-Ro and anti-La may be present, although Sharp’s criteria define anti Sm antibodies as a criterion for exclusion (Table 3).
Table 3. These are the investigations we undertake in patients with suspected MCTD
Haematology |
- FBC
- Coagulation profile including Lupus anticoagulant
- Direct Anti-globulin Test (DAT)
|
Biochemistry |
- Liver function
- Renal function
- Muscle enzymes
- Thyroid function
|
Inflammation |
|
Immunology |
- Immunoglobulins
- C3 and C4
|
Autoantibodies |
- RF
- ANA
- Anti-ds DNA
- Extractable Nuclear Antigens (ENA)
- Anti cardiolipin and anti beta2 glycoprotein antibodies
- Anti-neutrophil cytoplasmic antibodies (ANCA)
|
Imaging (as indicated) |
- Capillaroscopy
- X ray (chest)
- USS (hands, salivary glands, kidneys)
- MRI (joints, muscles)
- HRCT (chest)
|
Multi system screening |
- Urine protein: creatinine ratio
- ECG, echocardiogram, PFT
- Ophthalmology review
|
Histopathology (as indicated) |
- Biopsies – skin / renal / muscle
|
There is no specific treatment for MCTD and interventions (pharmacological and non-pharmacological) are directed at alleviating the symptoms. Most children with skin, joint and mild muscle involvement show initial response to low-dose corticosteroids, non-steroidal anti-inflammatory drugs (NSAIDs) and HCQ, or their combination [2,19]. RP is best managed by conservative measures, with only the most severe cases needing vasodilators. Patients with severe myositis and systemic involvement may need high dose corticosteroids and cytotoxic agents like Cyclophosphamide. The other agents used are MTX, Azathioprine, Cyclosporine, MMF, and biological agents like Etanercept and Infliximab. Rituximab has shown some encouraging results, especially in patients with thrombocytopaenia [21].
During the developmental period the course of the disease is milder in comparison to adults [9,10]. In majority of patients, after a few years the activity of the disease is low [8-10]. The most frequent causes of death are connected with development of pulmonary hypertension and interstitial lung disease [8-12].
Paediatric-onset MCTD carries less morbidity and mortality than adult-onset MCTD (because pulmonary hypertension is less common) [19]. Children have a favourable outcome 82% of the time, with disease characteristics of SLE and JDM/PM disappearing over time and SSc, arthritis, and RP persisting, but complete remission was rare [19]. Knight et al. reported that depression and anxiety symptoms, including suicidal ideation, were significantly more common in paediatric SLE/MCTD patients than in healthy subjects [22].
Childhood MCTD is a distinct but rare clinical entity which poses significant diagnostic challenge and can cause significant morbidity. A high index of suspicion and early referral to paediatric rheumatologist is recommended, as these patients need specialised input including multi system evaluation and multidisciplinary approach to management.
2021 Copyright OAT. All rights reserv
- Petty, Laxer, Lindsley, Wedderburn (2015) Mixed Connective Tissue Disease. Textbook of Pediatric Rheumatology, 7th Edition Elsevier 418- 426.
- Berard RA, Laxer RM (2016) Pediatric Mixed Connective Tissue Disease. Curr Rheumatol Rep 18: 28. [Crossref]
- Swart JF, Wulffraat NM (2008) Diagnostic workup for mixed connective tissue disease in childhood. Isr Med Assoc J 10: 650-652. [Crossref]
- Usuba FS, Lopes JB, Fuller R, Yamamoto JH, Alves MR, et al. (2014) Sjögren's syndrome: An underdiagnosed condition in mixed connective tissue disease. Clinics (Sao Paulo) 69: 158-162. [Crossref]
- Kotajima L (1996) Clinical features of patients with juvenile onset mixed connective tissue disease: analysis of data collected in a nationwide collaborative study in Japan. J Rheumatol 23: 1088-1094. [Crossref]
- Sumit Sen, Sinhamahapatra P, Choudhury S, Gangopadhyay A, Bala S, et al. (2014) Cutaneous Manifestations of Mixed Connective Tissue Disease: Study from a Tertiary Care Hospital in Eastern India. Indian J Dermatol 59: 35-40. [Crossref]
- Joyce Wang (2014) Unusual presentation of erythema multiforme in a patient with scleroderma/ mixed connective tissue disease. J Am Acad Dermatol 70: 57.
- Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR (1972) Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med 52: 148-159. [Crossref]
- Aringer M, Steiner G, Smolen JS (2005) Does mixed connective tissue disease exist? Yes. Rheum Dis Clin North Am 31: 411-420, v. [Crossref]
- Kahn MF, Appelboom T. In: Kahn MF, Peltier AP, Meyer O, et al. (1991) Les Maladies Systemiques. Paris: Flammarion. p. 545-556.
- Sharp GC. In: Kasukawa R, Sharp GC, editors (1987) Mixed connective tissue diseases and anti-nuclear antibodies. Amsterdam: ExcerptaMedica. p. 23-32.
- Kasukawa R. In: Kasukawa R, Sharp GC, editors (1987) Mixed connective tissue disease and anti-nuclear anti-bodies. Amsterdam: ExerptaMedica. 1987. p. 41-48.
- Alarcon-Segovia D, Villarreal M. In: Kasukawa R, Sharp GC, editors (1987) Mixed connective tissue disease and anti-nuclear anti-bodies. Amsterdam: ExcerptaMedica; 1987. p. 33-40.
- Amigues JM, Cantagrel A, Abbal M, Mazieres B (1996) Comparativestudy of 4 diagnosis criteria sets formixed connective tissue diseasein patients with anti-RNP antibodies. Autoimmunity Group of theHospitals of Toulouse. J Rheumatol 23: 2055-2062. [Crossref]
- Cappelli S (2012) BTo be or not to be,^ ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum 41:589-598.
- Distler JH, Strapatsas T, Huscher D, Dees C, Akhmetshina A, et al. (2011) Dysbalance of angiogenic and angiostatic mediators in patients with mixed connective tissue disease. Ann Rheum Dis 70: 1197-1202. [Crossref]
- Paradowska-Gorycka A (2015) Endosomal TLRs and their biological significance in the pathogenesis of mixed connective tissue disease. Reumatologia 3: 143-215.
- Racaza GZ (2014) Mixed connective tissue disease in Filipinos - A 13-year retrospective review of 14 cases in the Philippine general hospital: Phillippine. J Int Med 52
- Mier RJ, Shishov M, Higgins GC, Rennebohm RM, Wortmann DW, et al. (2005) Pediatric-onset mixed connective tissue disease. Rheum Dis Clin North Am 31: 483-496, vii. [Crossref]
- Aalokken TM, Lilleby V, Søyseth V, Mynarek G, Pripp AH, et al. (2009) Chest abnormalities in juvenile-onset mixed connective tissue disease: assessment with high-resolution computed tomography and pulmonary function tests. Acta Radiol 50: 430436. [Crossref]
- Jovancevic B, Lindholm C, Pullerits R (2013) Anti B-cell therapy against refractory thrombocytopenia in SLE and MCTD patients: long-term follow-up and review of the literature. Lupus 22: 664-674. [Crossref]
- Knight A, Pamela F. Weiss, Knashawn Morales, Ron Keren (2013) Depression, anxiety and suicidal thoughts in a cohort of pediatric lupus and mixed connective tissue disease patients. Arthritis and Rheumatism 65.