Abstract
The prevalence of overweight/obese adults and children continues to rise and with it the incidence of many secondary health consequences. Inflammation is pivotal in the manifestation of the comorbidities of obesity and is a product of dysregulated adipose tissue. Hence, obesity is a state of fat-induced prolonged inflammation that causes a decrease in immune defense. It is well established that central/visceral fat accumulation (the “apple” shape) is a risk factor for many adverse inflammatory metabolic outcomes associated with obesity, whereas peripheral or subcutaneous fat (the “pear” shape) is associated with a reduced risk. Considerable gains have been made in characterizing these regional differences in adipose tissue inflammation-induced metabolic dysregulation, but very little is known about the role of the immune system despite obesity being an inflammatory disease. The aim of this review is to elucidate how the lymphatic system, that drains tissues such as inflamed adipose tissue, may play a role in obesity-induced inflammation. Particular focus is placed on how the gastrointestinal tract that is connected to visceral adipose tissue via the lymphatic system may play a role in differential metabolic outcomes of distinct adipose tissue distributions. Overall, to accurately understand the pathogenic mechanisms that lead to the prolonged state of inflammation induced by obesity the role of the lymphatics must be elucidated.
Key words
obesity, inflammation, adipose, overweight
Introduction
Obesity is a growing epidemic. In 2014 the National Center for Health Statistics reported that ~70% of adults in the United States were overweight and that half of the overweight individuals were also obese [1]. Obesity, as traditionally measured by body mass index (BMI), is a risk factor for several diseases including, but not limited to, dyslipidemia, hypertension [2], non-alcoholic fatty liver disease (NAFLD) [3], atherosclerosis, cardiovascular disease (CVD) [4], and type-2-diabetes [5]. Consequently this condition is also a strong predictor of mortality [6]. However, exclusive markers of obesity such as positive energy balance and BMI are not accurate predictors of metabolic dysregulation. Rather adipose tissue distribution is the best determinant of an individual’s risk towards the development metabolic disease. It is common to investigate the differential aspects of adipose tissue distribution and its relation to metabolic risk via comparison of two different body types, the apple shape (visceral/central adipose tissue accumulation) or the pear (lower body subcutaneous adipose tissue distribution). Visceral adipose tissue accumulation occurs within the intra-abdominal cavity among vital organs such as the liver, pancreas, gut and kidneys whereas accumulation of lower body subcutaneous adipose tissue occurs between the muscle and skin (hypodermis) of the thighs, buttocks and lower stomach.
The concept that central obesity produces metabolic outcomes that are distinct from peripheral obesity is well established [7,8]. Numerous epidemiological studies demonstrate that excessive accumulation of visceral adipose tissue is associated with pathological conditions such as hypertension, dyslipidemia [9-11] (increased circulating triglycerides, free fatty acids, LDL to HDL ratio), cardiovascular disease, type-2 diabetes [12-16] and non-alcoholic fatty liver disease [17]. In opposition, studies propose that peripheral adipose tissue is protective, functioning as a “metabolic sink” that protects against lipid accumulation in non-adipose tissues. Consistent with this subcutaneous adipose tissue accumulation is associated with cardioprotection [18]. In particular, when compared with visceral adiposity, individuals with subcutaneous adiposity had lower circulating triglycerides and higher HDL cholesterol [18]. This relation between subcutaneous adipose tissue deposition and metabolic protection, however, is best associated with gluteofemoral subcutaneous adiposity. Truncal/abdominal subcutaneous adiposity is implicated in the pathogenesis of insulin resistance [19-21], atherosclerosis and cardiometabolic risk [22,23], whereas gluteofemoral adiposity is associated with insulin sensitivity, higher HDL and decreased risk for type-2-diabetes and metabolic syndrome [24-29]. We have begun to elucidate the mechanisms that causally relate the different metabolic outcomes of adipose tissue distribution [30-33] and have demonstrated that the strong link between visceral obesity and adverse metabolic outcomes is both anatomic location, proximity to the hepatic portal vein, and adipose depot specific physiology. Adipose depot specific characteristics, however, are also influenced by outside (extrinsic) factors such as, but not limited to, the immune (lymphatic) system. Hence below we discuss the postulate that the lymphatic system plays an important role in central/visceral obesity-mediated metabolic impairments. In addition, we will discuss how the gastrointestinal tract may prime visceral adiposity to be more detrimental than subcutaneous.
Adipose tissue
Visceral adipose tissue has the greatest association with pathogenesis of obesity-related disease despite its relatively low amount which constitutes 10% of total adiposity, whereas subcutaneous adipose tissue is ~85% [34]. Despite this the visceral adipose depot is proposed to play a deleterious role in obesity-induced health consequences because, as previously mentioned, where it is located. Hence, previous investigations primarily focus on visceral adipose tissue and liver interactions because effluent from visceral adipocytes containing metabolites, secretory products and inflammatory cytokines to the portal vein can be detrimental to insulin-sensitive hepatocytes [35-37]. These factors have major influence on hepatic processes such as glycogenesis, gluconeogenesis and very low-density lipoproteins (VLDL) synthesis [38-43]. Although the liver does play a large role in the exacerbation of obesity-induced metabolic disease, visceral adipose tissue surrounds, covers, attaches and shares vasculature with numerous other abdominal organs, yet these tissues are less investigated.
Adipose tissue is a complex endocrine organ with fundamental metabolic and immune regulatory roles. Adipocytes are characterized to release numerous hormones and signaling molecules, adipokines or adipocytokines that work in an autocrine and paracrine fashion or peripherally in an endocrine fashion. Adipocytokines regulate numerous physiological processes. In particular the role adipocytokines play in chronic pro-inflammation during obesity is gaining attention. Specifically, obesity associated adipocyte hypertrophy and hyperplasia dysregulates the sensitive microenvironment within adipose depots and alters their physiological processes, subsequently this can lead to changes in whole body homeostasis. Obesity-induced inflammation evoked during this process has been identified to be pivotal in the manifestation of the metabolic comorbidities and is identified to be a product of dysregulated adipose tissue. Hence, the short-term inflammation in adipose tissue that is a principal defense in response to acute injury becomes dysregulated during prolonged inflammation, as occurs in obesity, and is no longer beneficial [44]. Overall, obesity is a state of fat-induced prolongs inflammation.
Inflammation, induced by excessive adipose tissue accumulation, appears to link obesity to disease risk [45-47]. It is well established that central/visceral fat accumulation is a risk factor for many adverse inflammatory metabolic outcomes associated with obesity, whereas peripheral or subcutaneous fat is associated with a reduced risk. Hence, the increased propensity for individuals with visceral obesity to experience comorbidities appears to be linked to the increased capacity of this depot to induce inflammation [48]. Numerous studies demonstrate that visceral fat from obese individuals is characterized by increased macrophage infiltration, cytokine and chemokine production and other supportive immune cell types compared with non-visceral adipose tissue [49-53]. Despite considerable gains in characterization of regional differences in adipose tissue inflammation-induced metabolic dysregulation, very little is known about the role of the actual immune system in obesity-induced inflammation.
Lymphatic system and lymph nodes
The health and regulation of adipose tissue is primarily determined by the lymphatic system. Lymphatic system functions include; 1) interstitial fluid removal, 2) white blood cell transport, 3) absorption and transportation of lipids and 3) activation and initiation of immune cells and response. Overall, this system, which openly circulates throughout the body, is the conduit for immune cells that serves to produce an immune response to pathogen invasion and tissue injury. Lymph nodes are lymphatic system structures that are the primary site for the development of protective immune responses, including antibody responses and cellular immune responses that work via filtering and releasing immune cells to and from tissues. Regardless of location, lymph nodes are primarily characterized to occur embedded in adipose tissue depots [54]. Lymph nodes embedded in and attached to adipose tissue continuously survey and monitor exposure of adipose tissue to potentially harmful pathogens and metabolites [55,56]. Various immune cells within lymph nodes can be recruited and activated to defend adipose tissue against tissue damage, toxicity or impaired function [57]. Despite this association the relation between the adipose tissue and lymph node regulation is underemphasized. To accurately understand the pathogenic mechanisms that lead to the prolonged state of inflammation induced by obesity the role of the lymphatic system and adipose tissue depot crosstalk must be elucidated.
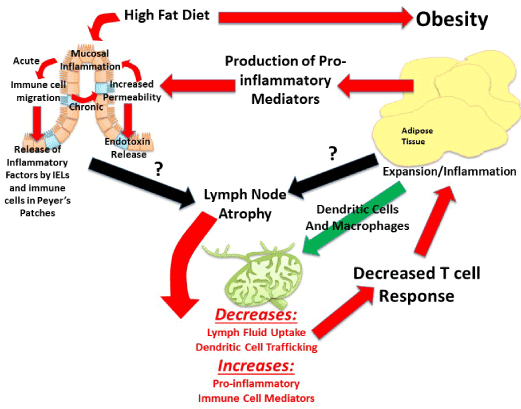
Though research is not yet extensive in lymph node and adipose tissue crosstalk, seminal studies indicate that obesity-induce immune dysregulation, chronic pro-inflammation, is likely due to fundamental alterations that occur in lymph node morphology, immune cell populations and immune response. Kim et al. demonstrate that the adverse inflammatory profile specific to visceral adipose tissue accumulation, but not subcutaneous, may be due to site-specific dysregulation of lymph nodes. First, they demonstrate that diet-induced adipose tissue accumulation reduces the weight of mesenteric, but not subcutaneous, lymph nodes [58]. The decrease in visceral lymph node mass is associated with a decrease in lymphoid cell number which is a result of activated T cell death, a measure the was ~4 fold higher in high fat diet fed mice than control [58]. This group proposed that various obesity-induced mediators (e.g. free fatty acids, oxidative stress, and inflammatory cytokines) that are released at a greater rate from mesenteric adipose tissue than subcutaneous cause greater immune cell apoptosis within this region [58]. They further propose, but do not tests, that metabolic endotoxemia-induced lymphocyte death, increases in circulating lipopolysaccharide (LPS) due to enhanced gut permeability, could mediate metabolic consequences associated with differential adiposity distributions. This topic, gut-adipose tissue crosstalk, will be discussed more below. Weitman et al. also provide studies that support the postulate that impairment of the lymphatic system may be involved in the pathology associated with obesity. They examine if obesity impairs lymphatic fluid transport and consequently dendritic cell migration. They analyzed lymphatic flow by microlymphangiography via dermal lymphatic vessels and determine lymphatic flow to be delayed ~3 fold in obese mice compared with lean [56]. In addition, overall uptake, drainage and transport of interstitial fluid is decreased in these obese mice [56]. Taken together these alterations are associated with lymphatic fluid stasis, a condition of inhibited lymph flow that subsequently exacerbates disease risk due to decreased immune defense.Consistent with this dendritic cell migration was decreased ~5 fold in obese mice [56], indicating that compared with lean animals obese mice are prone to dysregulated cell-mediated immunity because of disturbances between dendritic and T cell populations. This, in turn, can exacerbate the deleterious cycle of chronic low grade inflammation because a proper immune response cannot be produced if antigen-presenting cells (dendritic cells) cannot interact with T cells that tailor immune responses to those antigens. Unlike Kim et al. the Weitman group did not investigate regional differences in lymphatic flow, hence it remains unknown if these changes are exacerbated within the visceral cavity and play a role in the immune dysfunction associated with visceral obesity. However, Weitman et al. replicated that obese mice have smaller lymph nodes and further extended previous studies by establishing these smaller nodes are associated with abnormal morphology [56]. Specific mechanisms for obesity-induced lymph node atrophy remain unclear.
These above studies suggest that the lymphatic system, that drains tissues including, but not limited to, inflamed adipose tissue, plays a role in obesity-induced inflammation. These studies also suggeststhat diet-induced obesity can differentially influence lymph node regulation according to location.Diet-induced obesityaffects lymphoid tissues which, in turn, will also directly impact the development of immunity, including immune responses to pathogens, infections, cancer and vaccines in obese individuals.Specifically, the inability to alleviate inflammation, as occurs in obesity, has been show to play a role in chronic diseases such as such as type 2 diabetes [59], immune-related diseases [60-63], and reduced effectiveness of vaccines [64]. Obesity impairs the ability to mitigate inflammation, however, the factors involved in this impairment are poorly understood and to our knowledge, the interaction between obesity and lymphatic system immune responses has received very little attention in the field of obesity research.
Obesity is not only considered a precursor for a number of chronic diseases but is also associated with an increased risk for poorer prognosis in many immune-mediated conditions [60-63]. Numerous studies in humans demonstrate that obesity attenuates host defense [64-67], thus increasing susceptibility to infections. In particular, data collect during the 2009 influenza outbreak demonstrated that obese individuals were at greater risk for infection caused by morbidity and mortality [68]. Obese individuals are also characterized by an impaired responsiveness to influenza vaccination [64]. Hence, it is important to understand how the immune system, specifically the lymphatics, contributes to obesity-induced inflammation because it will begin to address why obese individuals are more susceptible to disease.
Gastrointestinal tract
Traditionally, inflammation originating from adipose tissue was thought to be the fundamental initiator of themetabolic disease associated with obesity,but emerging evidence suggests the intestines contributes greatly to the development of metabolic disease [69]. The small intestine is the first interface between the body and diet, consequently this region is the first exposed to excessive and detrimental nutrients ingested such asexcessive sugar and fat intakethat leads to increased adiposity. Because of this the gastrointestinal tract is now emerging as initiator of the events that contribute to obesity-associated systemic inflammation.The underlying mechanisms for this connection are currently being elucidated and thus far are contributed to Western diet-induced alterations in gut microbiota and epithelial barrier disruptions, increase gut permeability, that increase endotoxin release.
Reduced microbial diversity and altered bacteria phylum ratios are associated with the metabolic syndrome phentotype [70]. However, research is just now distinguishing if changes in gut microbiota prompt the low grade chronic inflammation associated with obesity or if alterations in gut microbiota are a secondary consequence of obesity. Research thus far demonstrates that gut microbiota contributes to obesity-induced inflammation via regulation of adipose depot stores. Specifically, the gut microbiotais a factor that regulates adipose tissue storage via enhance lipid release from the liver and adipocyte lipid uptake [71] as well as increased energy harvested from diet driven by microbiota derived metabolites [72]. Therefore, high fat diet-induced alterations ingut microbiota play a role in obesity-induced inflammation via exacerbation of adipose tissueaccumulation.In addition,emerging research demonstrates that high fat diet-induced alterations in gut microbial composition play a role in inflammation of the intestine that likely leads to inflammation associated with obesity. More specifically, obesity prone rats exhibit alterations in microbiota, explicitly an increase in enterobacteriales (lipopolysaccharide associated species), along with increases in activation of toll-like receptor 4 (TLR4) and intestinal inflammation compared with obese resistant rats [73]. The previous factors where demonstrated to be the necessary initiators for the development of the obese-prone phenotype [73]. This study and others [74] support the postulate that alterations in gut microbiota leading to increased intestinal inflammation and bacterial translocation are fundamental in the development of chronic low grade inflammation associated with obesity.
Overall, there is a strong link between high fat-induced alterations in gut microbiota, increases in harmful gut bacteria, gut inflammation and inflammation observed in obesity. Gut microbiota component lipopolysaccharide (LPS) an endotoxin, the main constituent of the outer membrane of Gram negative bacteria, plays a fundamental role in obesity-induced chronic low grade inflammation because it causes immune system activation. LPS is a major inducer of the inflammatory response and is increased in blood circulation following chronic fat ingestion because, as described above, diets high in fat cause increased gastrointestinal permeability and bacterial translocation (increased gut leakage) [75]. In obesity adipose tissue endotoxaemia triggers an innate response that causes the release of proinflammatory cytokines, hence exacerbating chronic low grade inflammation already produced by excessive adipose tissue accumulation.
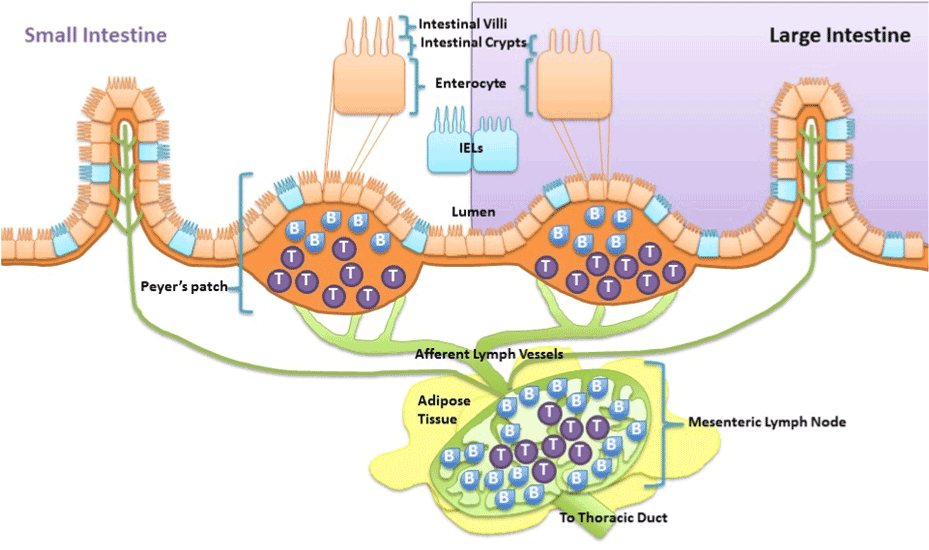
Taken together, obesity-induced chronic low grade inflammation development is attributed to and exacerbated by the deleterious cycle betweendysregulated gut and adipose tissue. As discussed above it is recognized that high fat diet-induced alterations in gut microbiota, altered gut metabolite release, gut inflammation and increased gastrointestinal permeability are fundamental drivers of the dysregulation. However, research suggests that high fat diet-induced intestinal inflammation does not just manifest chronically due to altered gut microbiota, but can also occur acutely due to direct effects of the diet. First, it has been demonstrated in rat epithelial cells that nutrient absorption of long chain fatty acids modulates intestinal mucosal immunity via enhancing cytokine release specific for neutrophil migration [76]. Thus excessive exposure of long chain fatty acids cause intestinal epithelial cells to release certain cytokines that cause migration of first responder phagocytes, neutrophils, to defend against the insult and activate other immune cells.Being persistently immunologically challenged, the epithelial cells of the gastrointestinal tract are integrated with immune cells. As such, the gut is characterized to contain 70% of the body’s immune cells which play a role in maintaining homeostasis of the gut [77]. These immune cells found along the epithelium of the intestine, intraepithelial lymphocytes (IEL), and within peyer’s patches, aggregated lymphoid nodules, release afferent lymph to the mesenteric lymph nodes that are surrounded by visceral adipose tissue (Figure 1). Much like intestinal epithelial cells, immune cells of the gastrointestinal tract are proposed to be capable of modifying immune function of the intestinal mucosa when directly exposed to fatty acids. In vitro long chain free fatty acids are demonstrated to alter interferon-Ɣ release from IELs collected from BALB/c mice [78] and suppress antigen presentation and chemotactic ability of dendritic cells [79]. In vivo fat absorption, specifically long chained fatty acids, in rats is demonstrated to activate intestinal mucosal mast cells and associated mediators [80]. In mice, a diet high in cholesterol causes accumulation of myeloid cells within the intestine within hours of exposure [81]. Overall, both intestinal mucosal epithelial cells and resident antigen cells are capable of directly detecting and inducing inflammatory responses to lipids such as cholesterol and long chain fatty acids.Because these intestinal immune populations drain to the mesenteric lymph nodes it islikely regional differences in lymph node immune cell can be driven by direct diet effect on the intestines and may play a role in the relation of adipose tissue distribution to metabolic disease.
Conclusion
Visceral adipose tissue surrounds, covers, attaches and shares vasculature with numerous abdominal organs, thus location of this depot is proposed to play a fundamental role in the co-morbidities associated with obesity, whereas subcutaneous adiposity is not. The increased link to obesity comorbidities appears to be due to the increased capacity of this depot to induce inflammation [48]. The next major advance in understanding the obesity epidemic will involve an appreciation that not all fat pads are created equally and an understanding of the fundamental processes that regulate inflammation. Visceral adipose tissue surrounds and attaches to the mesenteric lymph nodes that collect lymph fluid and immune cells effluent from the gastrointestinal tract. Although lymph nodes are the nexus of the gastrointestinal tract and visceral adipose tissue, its role is often underemphasized despite being fundamental. In particular, little is known about regional lymph node induction of adipose tissue inflammation. We postulate that obesity-mediated changes in lymph node immune cells within adipose depots determine the extent and negative consequences associated with obesity-induced inflammation. The contribution of intestinal immune cell populations and cytokine release to regional adiposity risk has yet to be investigated. The lymphatic connection between the gastrointestinal tract and visceral adipose tissue permits early diet direct immune alterations in the intestine to be detected within mesenteric lymph nodes. Resulting changes in mesenteric lymph nodes can directly affectthe adipose depot it resides in. Hence, high-fat diet that modulates immune functions in the intestinal mucosa produces gut-derived proinflammatory cytokines and associated immune cells, which will then directlycontribute to alterations in cell populations within the visceral adipose depot. This location specific lymph node regulation likely drives the differences between the associate metabolic risk of the “apple” and “pear” shape. Figure 2 is summary of proposed mechanisms of visceral obesity-induced chronic low grade inflammation.
References
- Health United States (2014) With Special Feature on Adults. Center for Disease Control and Prevention 55-64.
- Brown CD (2000) Body mass index and the prevalence of hypertension and dyslipidemia. Obes Res 8: 605-619. [Crossref]
- Miyake T (2013) Body mass index is the most useful predictive factor for the onset of nonalcoholic fatty liver disease: a community-based retrospective longitudinal cohort study. J Gastroenterol 48: 413-22. [Crossref]
- Lamon-Fava S, Wilson PW, Schaefer EJ (1996) Impact of body mass index on coronary heart disease risk factors in men and women. The Framingham Offspring Study. Arterioscler Thromb Vasc Biol 16: 1509-1515. [Crossref]
- Sanada H, Yokokawa H, Yoneda M, Yatabe J, Sasaki Yatabe M, et al. (2012) High body mass index is an important risk factor for the development of type 2 diabetes. Intern Med 51: 1821-1826. [Crossref]
- Haffner SM (2006) Relationship of metabolic risk factors and development of cardiovascular disease and diabetes. Obesity (Silver Spring) 14 Suppl 3: 121S-127S. [Crossref]
- Després JP, Lemieux S, Lamarche B, Prud'homme D, Moorjani S, et al. (1995) The insulin resistance-dyslipidemic syndrome: contribution of visceral obesity and therapeutic implications. Int J Obes Relat Metab Disord 19 Suppl 1: S76-86. [Crossref]
- Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, et al. (2007) Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 116: 39-48. [Crossref]
2021 Copyright OAT. All rights reserv
- Mamo JC, Watts GF, Barrett PH, Smith D, James AP, et al. (2001) Postprandial dyslipidemia in men with visceral obesity: an effect of reduced LDL receptor expression? Am J Physiol Endocrinol Metab 281: E626-632. [Crossref]
- Chan DC, Barrett HP, Watts GF (2004) Dyslipidemia in visceral obesity: mechanisms, implications, and therapy. Am J Cardiovasc Drugs 4: 227-246. [Crossref]
- Zambon A, Marchiori M, Manzato E (2008) Dyslipidemia in visceral obesity: pathophysiological mechanisms, clinical implications and therapy. G Ital Cardiol (Rome) 9: 29S-39S. [Crossref]
- Anjana M, Sandeep S, Deepa R, Vimaleswaran KS, Farooq S, et al. (2004) Visceral and central abdominal fat and anthropometry in relation to diabetes in Asian Indians. Diabetes Care 27: 2948-2953. [Crossref]
- Hayashi T, Boyko EJ, Leonetti DL, McNeely MJ, Newell-Morris L, et al. (2003) Visceral adiposity and the risk of impaired glucose tolerance: a prospective study among Japanese Americans. Diabetes Care 26: 650-655. [Crossref]
- Wei M (1997) Waist circumference as the best predictor of noninsulin dependent diabetes mellitus (NIDDM) compared to body mass index, waist/hip ratio and other anthropometric measurements in Mexican Americans--a 7-year prospective study. Obes Res 195: 16-23. [Crossref]
- Dowse GK, Zimmet PZ, Gareeboo H, George K, Alberti MM, et al. (1991) Abdominal obesity and physical inactivity as risk factors for NIDDM and impaired glucose tolerance in Indian, Creole, and Chinese Mauritians. Diabetes Care 14: 271-282. [Crossref]
- Araneta MR, Barrett-Connor E (2005) Ethnic differences in visceral adipose tissue and type 2 diabetes: Filipino, African-American, and white women. Obes Res 13: 1458-1465. [Crossref]
- Speliotes EK, Massaro JM, Hoffmann U, Vasan RS, Meigs JB, et al. (2010) Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: the Framingham Heart Study. Hepatology 51: 1979-1987. [Crossref]
- Porter SA, Massaro JM, Hoffmann U, Vasan RS, O'Donnel CJ, et al. (2009) Abdominal subcutaneous adipose tissue: a protective fat depot? Diabetes Care 32: 1068-1075. [Crossref]
- Abate N, Garg A, Peshock RM, Stray-Gundersen J, Grundy SM (1995) Relationships of generalized and regional adiposity to insulin sensitivity in men. J Clin Invest 96: 88-98. [Crossref]
- Goodpaster BH, Thaete FL, Simoneau JA, Kelley DE (1997) Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes 46: 1579-1585. [Crossref]
- Abate N, Garg A, Peshock RM, Stray-Gundersen J, Adams-Huet B, et al. (1996) Relationship of generalized and regional adiposity to insulin sensitivity in men with NIDDM. Diabetes 45: 1684-1693. [Crossref]
- Ferreira (2005) The metabolic syndrome, cardiopulmonary fitness, and subcutaneous trunk fat as independent determinants of arterial stiffness: the Amsterdam Growth and Health Longitudinal Study. Arch Intern Med 165: 875-882. [Crossref]
- Wildman RP, Janssen I, Khan UI, Thurston R, Barinas-Mitchell E, et al. (2011) Subcutaneous adipose tissue in relation to subclinical atherosclerosis and cardiometabolic risk factors in midlife women. Am J Clin Nutr 93: 719-726. [Crossref]
- Snijder MB, Visser M, Dekker JM, Goodpaster BH, Harris TB, et al. (2005) Low subcutaneous thigh fat is a risk factor for unfavourable glucose and lipid levels, independently of high abdominal fat. The Health ABC Study. Diabetologia 48: 301-308. [Crossref]
- McLaughlin (2011) Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J Clin Endocrinol Metab 96: E1756-1760. [Crossref]
- Koster A, Stenholm S, Alley DE, Kim LJ, Simonsick EM, et al. (2010) Body fat distribution and inflammation among obese older adults with and without metabolic syndrome. Obesity (Silver Spring) 18: 2354-2361. [Crossref]
- Gallagher D, Kelley DE, Yim JE, Spence N, Albu J, et al. (2009) Adipose tissue distribution is different in type 2 diabetes. Am J Clin Nutr 89: 807-814. [Crossref]
- Yim JE, Heshka S, Albu JB, Heymsfield S, Gallagher D (2008) Femoral-gluteal subcutaneous and intermuscular adipose tissues have independent and opposing relationships with CVD risk. J Appl Physiol (1985) 104: 700-707. [Crossref]
- Manolopoulos KN, Karpe F, Frayn KN (2010) Gluteofemoral body fat as a determinant of metabolic health. Int J Obes (Lond) 34: 949-959. [Crossref]
- Foster MT, Shi H, Seeley RJ, Woods SC (2010) Transplantation or removal of intra-abdominal adipose tissue prevents age-induced glucose insensitivity. Physiol Behav 101: 282-288. [Crossref]
- Foster MT, Shi H, Seeley RJ, Woods SC (2011) Removal of intra-abdominal visceral adipose tissue improves glucose tolerance in rats: role of hepatic triglyceride storage. Physiol Behav 104: 845-854. [Crossref]
- Foster MT, Shi H, Softic S, Kohli R, Seeley RJ, et al. (2011) Transplantation of non-visceral fat to the visceral cavity improves glucose tolerance in mice: investigation of hepatic lipids and insulin sensitivity. Diabetologia 54: 2890-2899. [Crossref]
- Foster MT, Softic S, Caldwell J, Kohli R, de Kloet AD, et al. (2013) Subcutaneous Adipose Tissue Transplantation in Diet-Induced Obese Mice Attenuates Metabolic Dysregulation While Removal Exacerbates It. Physiol Rep [Crossref]
- Klein S et al. (2007) Waist circumference and cardiometabolic risk: a consensus statement from shaping America's health: Association for Weight Management and Obesity Prevention; NAASO, the Obesity Society; the American Society for Nutrition; and the American Diabetes Association. Diabetes Care 30: 1647-1652. [Crossref]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, et al. (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271: 665-668. [Crossref]
- Bergman RN (2000) Non-esterified fatty acids and the liver: why is insulin secreted into the portal vein? Diabetologia 43: 946-952. [Crossref]
- Williamson JR, Kreisberg RA, Felts PW (1966) Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci U S A 56: 247-254. [Crossref]
- Nemecz M, Preininger K, Englisch R, Fürnsinn C, Schneider B, et al. (1999) Acute effect of leptin on hepatic glycogenolysis and gluconeogenesis in perfused rat liver. Hepatology 29: 166-172. [Crossref]
- Borba-Murad GR, Mario EG, Bassoli BK, Bazotte RB, de Souza HM (2005) Comparative acute effects of leptin and insulin on gluconeogenesis and ketogenesis in perfused rat liver. Cell Biochem Funct 23: 405-413. [Crossref]
- Bassil MS, Mrayati MM, Hwalla NC, Obeid OA (2007) Acute effect of leptin and ghrelin injection on postprandial glycogen and lipid synthesis in rats. Ann Nutr Metab 51: 14-21. [Crossref]
- Gerner RR, Wieser V, Moschen AR, Tilg H (2013) Metabolic inflammation: role of cytokines in the crosstalk between adipose tissue and liver. Can J Physiol Pharmacol 91: 867-872. [Crossref]
- Neuschwander-Tetri BA (2010) Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52: 774-788. [Crossref]
- Topping DL, Mayes PA (1982) Insulin and non-esterified fatty acids. Acute regulators of lipogenesis in perfused rat liver. Biochem J 204: 433-439. [Crossref]
- Mraz M, Haluzik M (2014) The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol 222: R113-127. [Crossref]
- Thijssen E, van Caam A, van der Kraan PM2 (2015) Obesity and osteoarthritis, more than just wear and tear: pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity-induced osteoarthritis. Rheumatology (Oxford) 54: 588-600. [Crossref]
- Guarner V, Rubio-Ruiz ME (2015) Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip Top Gerontol 40: 99-106. [Crossref]
- Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N (2014) Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract 105: 141-150. [Crossref]
- Ghigliotti G, Barisione C, Garibaldi S, Fabbi P, Brunelli C, et al. (2014) Adipose tissue immune response: novel triggers and consequences for chronic inflammatory conditions. Inflammation 37: 1337-1353. [Crossref]
- Cartier A (2010) Plasma soluble tumour necrosis factor-alpha receptor 2 is elevated in obesity: specific contribution of visceral adiposity. Clin Endocrinol (Oxf) 72: 349-357. [Crossref]
- Fried SK, Bunkin DA, Greenberg AS (1998) Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83: 847-850. [Crossref]
- Lemieux I, Pascot A, Prud'homme D, Alméras N, Bogaty P, et al. (2001) Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol 21: 961-967. [Crossref]
- Malavazos AE, Corsi MM, Ermetici F, Coman C, Sardanelli F, et al. (2007) Proinflammatory cytokines and cardiac abnormalities in uncomplicated obesity: relationship with abdominal fat deposition. Nutr Metab Cardiovasc Dis 17: 294-302. [Crossref]
- Curat CA, Wegner V, Sengenès C, Miranville A, Tonus C, et al. (2006) Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 49: 744-747. [Crossref]
- Pond CM, Mattacks CA (1995) Interactions between adipose tissue around lymph nodes and lymphoid cells in vitro. J Lipid Res 36: 2219-2231. [Crossref]
- Arngrim N, et al. (2013) Reduced adipose tissue lymphatic drainage of macromolecules in obese subjects: a possible link between obesity and local tissue inflammation? Int J Obes (Lond) 37: 748-750. [Crossref]
- Weitman ES, Aschen SZ, Farias-Eisner G, Albano N, Cuzzone DA, et al. (2013) Obesity impairs lymphatic fluid transport and dendritic cell migration to lymph nodes. PLoS One 8: e70703. [Crossref]
- von der Weid PY, Rainey KJ (2010) Review article: lymphatic system and associated adipose tissue in the development of inflammatory bowel disease. Aliment Pharmacol Ther 32: 697-711. [Crossref]
- Kim CS, Lee SC, Kim YM, Kim BS, Choi HS, et al. (2008) Visceral fat accumulation induced by a high-fat diet causes the atrophy of mesenteric lymph nodes in obese mice. Obesity (Silver Spring) 16: 1261-1269. [Crossref]
- Kissebah AH, Krakower GR (1994) Regional adiposity and morbidity. Physiol Rev 74: 761-811. [Crossref]
- Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y (2014) Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev 13: 981-1000. [Crossref]
- Kim SI, Kim HS, Kim TH, Suh DH, Kim K, et al. (2014) Impact of underweight after treatment on prognosis of advanced-stage ovarian cancer. J Immunol Res 2014: 349546. [Crossref]
- Schmitz KH, Neuhouser ML, Agurs-Collins T, Zanetti KA, Cadmus-Bertram L, et al. (2013) Impact of obesity on cancer survivorship and the potential relevance of race and ethnicity. J Natl Cancer Inst 105: 1344-1354. [Crossref]
- Novosad S, Khan S, Wolfe B, Khan A (2013) Role of obesity in asthma control, the obesity-asthma phenotype. J Allergy (Cairo): 538642. [Crossref]
- Sheridan PA, Paich HA, Handy J, Karlsson EA, Hudgens MG, et al. (2012) Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes (Lond) 36: 1072-1077. [Crossref]
- Milner JJ, Beck MA (2012) The impact of obesity on the immune response to infection. Proc Nutr Soc 71: 298-306. [Crossref]
- Bochicchio GV, Joshi M, Bochicchio K, Nehman S, Tracy JK, et al. (2006) Impact of obesity in the critically ill trauma patient: a prospective study. J Am Coll Surg 203: 533-538. [Crossref]
- Yaegashi M, Jean R, Zuriqat M, Noack S, Homel P (2005) Outcome of morbid obesity in the intensive care unit. J Intensive Care Med 20: 147-154. [Crossref]
- Mancuso P (2013) Obesity and respiratory infections: does excess adiposity weigh down host defense? Pulm Pharmacol Ther 26: 412-419. [Crossref]
- de Wit NJ, Bosch-Vermeulen H, de Groot PJ, Hooiveld GJ, Bromhaar MM, et al. (2008) The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med Genomics 1: 14. [Crossref]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, et al. (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102: 11070-11075. [Crossref]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, et al. (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101: 15718-15723. [Crossref]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, et al. (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027-1031. [Crossref]
- de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, et al. (2010) Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299: G440-448. [Crossref]
- Fei N, Zhao L (2013) An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J 7: 880-884. [Crossref]
- Teixeira TF, Collado MC, Ferreira CL, Bressan J, Peluzio Mdo C (2012) Potential mechanisms for the emerging link between obesity and increased intestinal permeability. Nutr Res 32: 637-647. [Crossref]
- Yoshida H, Miura S, Kishikawa H, Hirokawa M, Nakamizo H, et al. (2001) Fatty acids enhance GRO/CINC-1 and interleukin-6 production in rat intestinal epithelial cells. J Nutr 131: 2943-2950. [Crossref]
- Castro GA, Arntzen CJ (1993) Immunophysiology of the gut: a research frontier for integrative studies of the common mucosal immune system. Am J Physiol 265: G599-G610. [Crossref]
- Hara Y. et al, (2003) Exposure to fatty acids modulates interferon production by intraepithelial lymphocytes. Immunol Lett 86: 139-148. [Crossref]
- Tsuzuki Y, Miyazaki J, Matsuzaki K, Okada Y, Hokari R, et al. (2006) Differential modulation in the functions of intestinal dendritic cells by long- and medium-chain fatty acids. J Gastroenterol 41: 209-216. [Crossref]
- Ji Y, Sakata Y, Yang Q, Li X, Xu M, et al. (2012) Activation of rat intestinal mucosal mast cells by fat absorption. Am J Physiol Gastrointest Liver Physiol 302: G1292-1300. [Crossref]
- Progatzky F, Sangha NJ, Yoshida N, McBrien M, Cheung J, et al. (2014) Dietary cholesterol directly induces acute inflammasome-dependent intestinal inflammation. Nat Commun 5: 5864. [Crossref]