Abstract
Backgrounds underlying the age-related bone loss can be classified into two categories: systemic abnormality and osteoblast dysfunction. The former includes insufficiency of vitamin D or estrogen, causing a negative balance of calcium metabolism. We propose the contribution of an ageing-suppressing gene, klotho, as a novel systemic factor, since the mouse deficient in the klotho gene exhibits multiple aging phenotypes including osteopenia with a low bone turnover. As a factor intrinsic to osteoblasts, we investigated the role of PPARγ, a key regulator of adipocyte differentiation, based on the facts that osteoblasts and adipocytes share a common progenitor. Heterozygous PPARγ-deficient mice exhibited high bone mass by stimulating osteoblastogenesis from bone marrow progenitors, and this effect became prominent with ageing, indicating involvement of PPARγ-dependent bone formation in the pathophysiology of age-related bone loss. The local environment of osteoblasts is mainly controlled by cytokines/growth factors, among which insulin-like growth factor-I (IGF-I) is the most possible candidate whose production and activity are decreased with ageing. Bone phenotypes of deficient mice of insulin receptor substrates (IRS-1 and IRS-2), essential molecules for intracellular signaling of IGF-I, revealed that IRS-1 is essential to maintain bone turnover by up-regulating anabolic and catabolic functions of osteoblasts, while IRS-2 is needed to keep the predominance of the anabolic function over the catabolic function. A next task ahead of us will be to elucidate the network system of these factors underlying the age-related osteoporosis.
Key words
osteoporosis, klotho, pparγ, insulin receptor substrate (IRS)
Three major backgrounds of osteoporosis
There are three major backgrounds of osteoporosis in aged women: 1) the peak bone mass during their adolescence was low, 2) the bone loss by menopause due to estrogen deficiency was severe, and 3) the bone loss by ageing thereafter was severe (Figure 1); each of these has an independent mechanism. Regarding the mechanisms underlying the bone loss by ageing, accumulated evidence has suggested many age-related abnormalities which can be classified into two categories: systemic abnormality and osteoblast dysfunction (Figure 2). The former includes insufficiency of active vitamin D or estrogen, which decreases calcium absorption from G.I. and kidney, causing a negative balance of calcium metabolism and a secondary hyperparathyroidism [1-8]. The latter can be further divided into abnormalities that occur inside and outside of osteoblasts. As an inside factor, Runx2, a key transcription factor for osteoblast differentiation, is the most probable candidate, since the expression is reported to be suppressed during cellular ageing of osteoblasts [9]; however, there is no in vivo evidence of its contribution to age-related bone loss. The local environment outside osteoblasts is mainly controlled by cytokines/growth factors such as insulin-like growth factor-I (IGF-I) [10-12], interleukin-11 [13], transforming growth factor-β [14] and bone morphogenetic proteins [15].
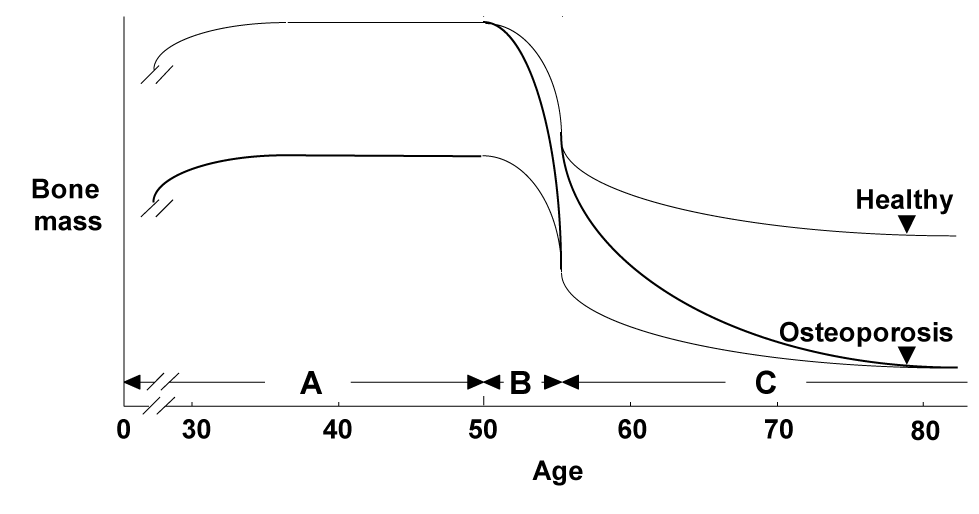
Figure 1. Three major backgrounds for the etiology of osteoporosis.
A: The peak bone mass in the adolescence. B: A rapid bone loss after menopause. C: A gradual age-related bone loss thereafter.
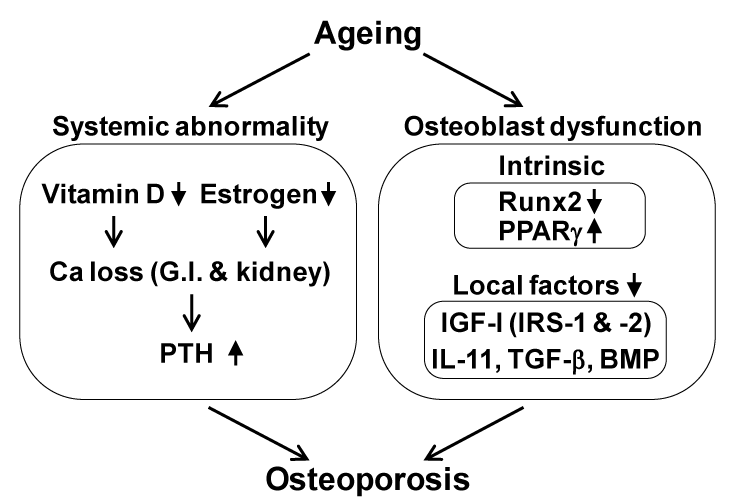
Figure 2. Possible mechanisms underlying bone loss by ageing.
IL-11, interleukin-11; TGF-β, transforming growth factor-β; BMP, bone morphogenetic protein; G.I., gastrointestinal; PTH, parathyroid hormone
None of these hormones, cytokines, or transcription factor, however, can fully explain the etiology of age-related bone loss. To further investigate its molecular backgrounds, we have been involved in the reverse and forward genetic approaches: the reverse genetics is the approach from a genotype to a phenotype using gene-manipulated mice such as knocked-out or transgenic, while the forward genetic approach is from a disease to the responsible gene using the human genomic analysis, based on the reverse genetic findings. This review summarizes the possible roles of three different types of molecules, a hormone klotho, an osteoblast intrinsic factor peroxisome proliferator-activated receptor-γ (PPARγ), and a local factor IGF-I by way of its adaptor molecule insulin receptor substrates (IRS), in age-related bone loss primarily from our recent mouse genetics approaches.
Klotho as a hormone in age-related osteoporosis
In addition to hormones vitamin D and estrogen, we hereby propose the contribution of an ageing-suppressing gene, klotho, as a novel systemic factor regulating age-related bone loss. Klotho was originally identified as a mutated gene in a mouse strain that accelerates age-dependent loss of function in multiple age sensitive traits [16]. An insertional mutation that disrupts the 5’promoter region of the klotho gene resulted in a strong hypomorphic allele. Mice homozygous for the mutated allele (KL-/- mice) exhibited multiple age-related disorders including osteopenia, especially in the cortex bones, just like human senile osteoporosis (Figure 3A), and suffered premature death around two months of age [17,18]. Bone histomorphometric analysis revealed that parameters of both bone formation (Ob.S/BS and BFR/BS) and bone resorption (Oc.N/B.Pm and ES/BS) were lower in KL-/- mice in the wild-type (WT) littermates, with predominant decreases of the formation parameters over the resorption parameters (Figure 3B), indicating a state of low bone turnover osteopenia.
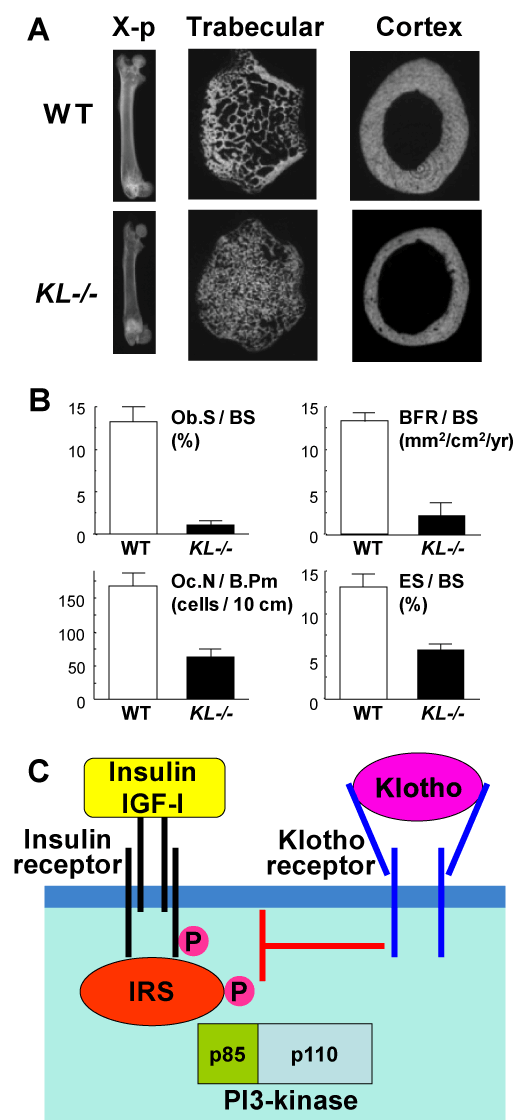
Figure 3. Bone phenotype of KL-/-mice (A & B) and interaction between klotho and insulin / IGF-I signalings (C)
(A) Plain X-ray and CT images of femora of KL-/- and WT littermates at 8 weeks of age. (B) Bone histomorphometric analysis of the proximal tibiae of KL-/- and WT littermates. Ob.S / BS, osteoblast surface per bone surface; BFR / BS, bone formation rate per bone surface; Oc.N / B.Pm, osteoclast number per bone perimeter; ES/BS, eroded surface per bone surface. (C) A scheme of the interaction between klotho and insulin / IGF-I signalings.
The klotho gene encodes a single-pass transmembrane protein that is detectable in limited tissues, particularly the distal convoluted tubules in the kidney and the choroid plexus in the brain. Because a defect in the klotho gene leads to systemic age-dependent degeneration, the klotho protein may function through a circulating humoral factor that regulates the development of age-related disorders or natural ageing processes [19]. We recently showed that overexpression of klotho can extend life span, and found that the extracellular domain of the klotho protein circulates in the blood and binds to a putative cell-surface receptor [20]. Klotho functions as a hormone that suppresses tyrosine phosphorylation of insulin and IGF-I (Figure 3C). Since extended life span upon negative regulation of insulin and IGF-I signaling is an evolutionarily conserved mechanism to suppress ageing [21], klotho appears to be a peptide hormone to modulate such signaling and thereby mediate insulin metabolism and ageing.
As the forward genetic approach, we examined the association between human klotho gene polymorphisms and bone density in postmenopausal women of two genetically distinct racial populations: the Caucasian and the Japanese. Screening of single-nucleotide polymorphisms (SNPs) in the human klotho gene identified a total of 11 SNPs, and three of them were common in both populations. Among them, two SNPs: one in the promoter region and one in exon 4 were significantly associated with bone density of the aged postmenopausal women in both populations [22]. Other SNPs in the human klotho gene are reported to be associated not only with bone loss [23,24], but also with altered life span [25] and risk for coronary artery disease [26] and stroke [27]. These results indicate that the klotho gene may be involved in the longevity and pathophysiology of age-related disorders including osteoporosis in humans.
PPARγ as an osteoblast intrinsic molecule in age-related osteoporosis
Osteoblasts and adipocytes are known to share a common progenitor: multipotential mesenchymal stem cells in bone marrow, being driven by respective key molecules Runx2 and PPARγ [28-31] (Figure 4A). In addition, ageing is associated with a reciprocal decrease of osteogenesis and an increase of adipogenesis in bone marrow [32-34] (Figure 4B). Based on these facts, we investigated the physiological role of PPARγ in bone metabolism [35], using heterozygous PPARγ-deficient (PPARγ+/-) mice [36]. Although they showed no abnormalities in major organs such as brain, heart, liver, spleen or kidney on a standard diet [36,37], they exhibited high bone mass both at young and old ages (Figure 5A). The time course of the bone volume revealed that bone mass was decreased with ageing in both PPARγ+/- and WT littermates; however, the difference of bone volume between the two genotype mice became more prominent at 52 weeks (Figure 5B), indicating the involvement of the PPARγ signaling in the pathophysiology of human age-related osteoporosis [35]. In fact, a previous association study between bone density and a genetic polymorphism of PPARγ in postmenopausal women implies the involvement of PPARγ in bone loss [38]. Ex vivo culture of bone marrow cells derived from PPARγ+/- and WT showed that PPARγ haploinsufficiency caused not only a decrease in the number of adipocytes, but also an increase of osteoblasts, indicating that PPARγ signaling in marrow progenitors functions as a potent suppressor of commitment to osteoblastic lineage [35].
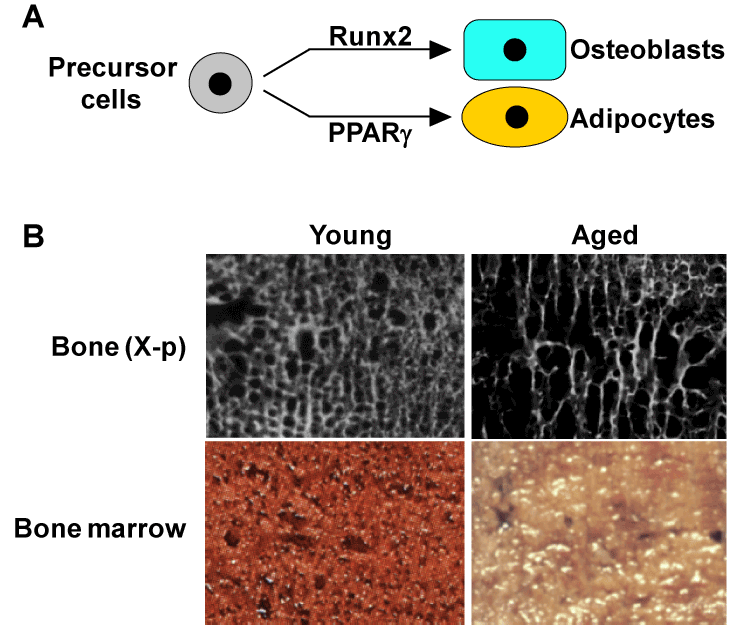
Figure 4. Backgrounds for possible involvement of PPARγ in age-related bone loss
(A) Mesenchymal precursor cells in bone marrow can differentiate into both osteoblasts and adipocytes through respective key molecules Runx2 and PPARγ. (B) X-ray (top) and normal pictures (bottom) of bone marrow in proximal femurs of young (34 yrs.) and aged (86 yrs.) females, both of whom underwent surgical operations due to fracture. In the aged marrow, a reciprocal decrease of bone mass and an increase of adipose tissue.
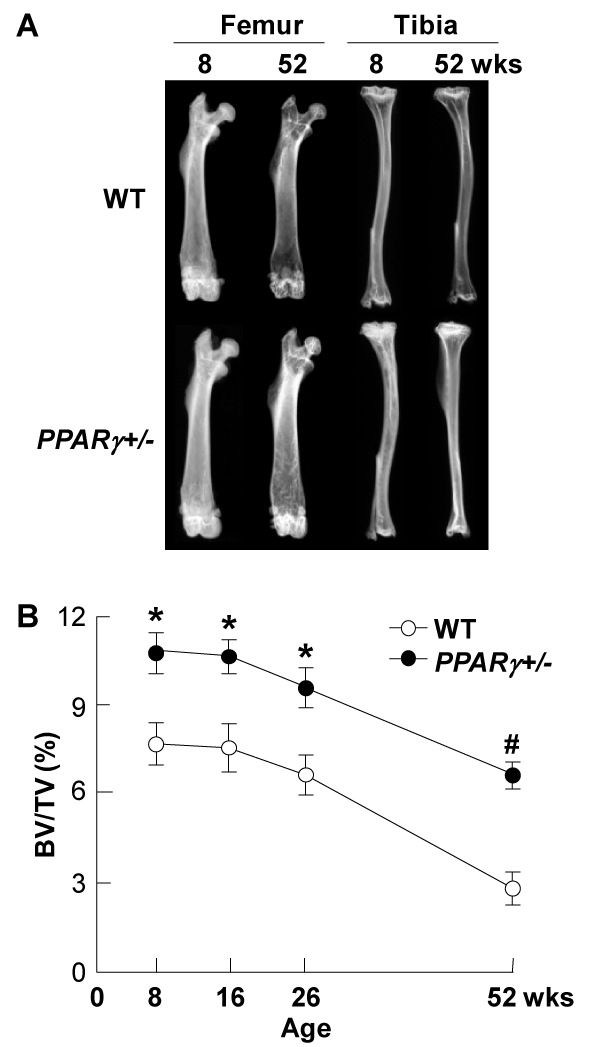
Figure 5. Radiological analyses of PPARγ+/- and WT littermates at indicated ages
(A) Plain X-ray images of femora and tibiae of representative PPARγ+/- and WT littermates at 8 and 52 weeks of age. (B) Time course of trabecular bone volume expressed as percentage of total tissue volume (BV/TV, %) at the distal femora was measured on the CT image. Data are expressed as means (symbols) ± SEMs (error bars) for 8mice / group for PPARγ+/- and WT. Significant difference from WT; *P<0.05, #P<0.01, determined by post-hoc testing using Bonferroni's method.
In addition to the role of PPARγ as an intracellular molecule, a recent report suggested that PPARγ could have an antiosteogenic endocrine role, since severely lipodystrophic PPARγhyp/hyp mice which have a hypomorphic mutation at the PPARγ locus in white adipose tissue [39] showed enhanced bone formation [40]. Adipokines, such as leptin and adiponectin, which are secreted by adipocytes and potently regulate bone metabolism [41-45] might be involved in the PPARγ-related systemic signaling in bone formation.
IRS signaling in age-related osteoporosis
Among cytokines/growth factors of which decreases with ageing have been indicated to be responsible for osteoblast dysfunction, IGF-I is the most probable candidate whose serum level is decreased with ageing and positively related to bone density of aged populations [10-12]. IGF-I initiates cellular responses by binding to its cell-surface receptor tyrosine kinase IGF-I receptor, which then activates essential adaptor molecule IRS followed by downstream signaling pathways like phosphatidylinositol-3 kinase (PI3K)/Akt and mitogen-activated protein kinases (MAPKs) [46]. The mammalian IRS family contains at least four members: ubiquitous IRS-1 and IRS-2, adipose tissue-predominant IRS-3, and IRS-4 which is expressed in the thymus, brain and kidney. We previously reported that IRS-1 and IRS-2 are expressed in bone [47,48]. Our further studies on mice lacking the IRS-1 gene (IRS-1-/- mice) or the IRS-2 gene (IRS-2-/- mice) revealed that these knockout mice exhibited severe osteopenia with distinct mechanisms: IRS-1-/- mice showed a low bone turnover in which both bone formation and resorption were decreased [47], whereas IRS-2-/- mice showed an uncoupling status with decreased bone formation and increased bone resorption [48]. It therefore seems that under physiological conditions IRS-1 is important for maintaining bone turnover, while IRS-2 for retaining the predominance of anabolic function over catabolic function of osteoblasts. IGF-I may up-regulate bone formation without affecting bone resorption through the balance of the two signalings (Figure 6).
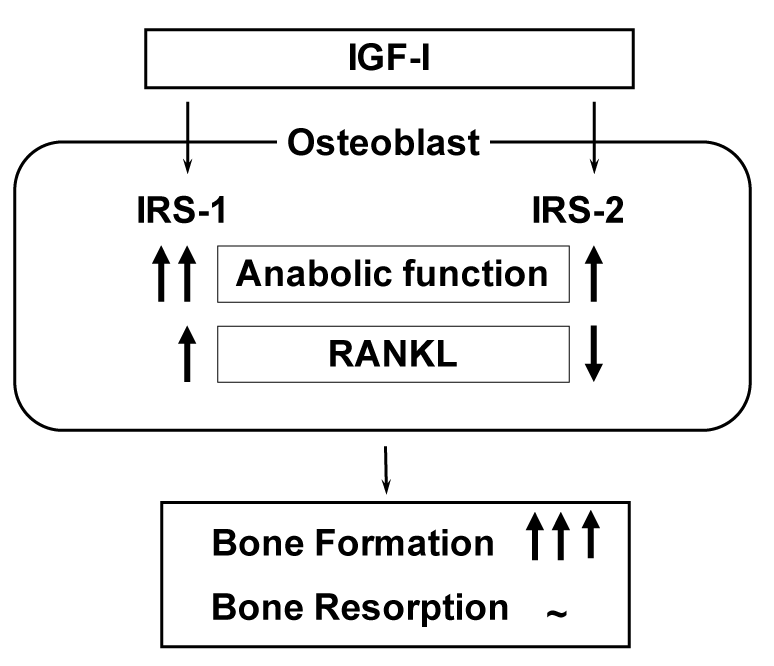
Figure 6. Mechanism of bone formation byIGF-I through IRS-1 and IRS-2 signalings.
Analyses of bones of IRS-1-/- and IRS-2-/- mice revealed that IRS-1 is important for maintaining both bone anabolic function and catabolic function through RANKL expression in osteoblasts, while IRS-2 increases the anabolic function but decreases the catabolic function. As a balance of the two signalings, IGF-I up-regulates bone formation without affecting bone resorption.
Conclusion
We hereby propose new players, klotho, PPARγ, and IGF-I through IRS-1 and IRS-2 signalings, in the mechanism of age-related osteoporosis, using mouse genetics approaches. These signalings may constitute a network with other molecules like vitamin D, estrogen, Runx2, other cytokines, etc. to maintain bone mass. In addition, there may be a complex interaction among the signalings. For example, considering that the insulin/IGF-I/IRS signaling exhibits a bone anabolic action as described above, the inhibitory action of klotho on the insulin/IGF-I/IRS signaling is inconsistent with the osteopenic phenotype of KL-/- mice. We believe that there are signal pathways other than the insulin pathway for the klotho action on bone metabolism. One of the next tasks ahead of us will be to elucidate the network system of these many factors involved in age-related bone loss.
References
- Bullamore JR, Wilkinson R, Gallagher JC, Nordin BE, Marshall DH (1970) Effect of age on calcium absorption. Lancet 2: 535-537. [Crossref]
- Gallagher JC, Riggs BL, Eisman J, Hamstra A, Arnaud SB, et al. (1979) Intestinal calcium absorption and serum vitamin D metabolites in normal subjects and osteoporotic patients: effect of age and dietary calcium. J Clin Invest 64: 729-736. [Crossref]
- Horst RL, Goff JP, Reinhardt TA (1990) Advancing age results in reduction of intestinal and bone 1,25-dihydroxyvitamin D receptor. Endocrinology 126: 1053-1057. [Crossref]
- Wood RJ, Fleet JC, Cashman K, Bruns ME, Deluca HF (1998) Intestinal calcium absorption in the aged rat: evidence of intestinal resistance to 1,25(OH)2 vitamin D. Endocrinology 139: 3843-3848. [Crossref]
- Slovik DM, Adams JS, Neer RM, Holick MF, Potts JT Jr (1981) Deficient production of 1,25-dihydroxyvitamin D in elderly osteoporotic patients. N Engl J Med 305: 372-374. [Crossref]
- Ledger GA, Burritt MF, Kao PC, O'Fallon WM, Riggs BL, et al. (1994) Abnormalities of parathyroid hormone secretion in elderly women that are reversible by short term therapy with 1,25-dihydroxyvitamin D3. J Clin Endocrinol Metab 79: 211-216. [Crossref]
- Haden ST, Brown EM, Hurwitz S, Scott J, El-Hajj Fuleihan G (2000) The effects of age and gender on parathyroid hormone dynamics. Clin Endocrinol (Oxf) 52: 329-338. [Crossref]
- Cummings SR, Browner WS, Bauer D, Stone K, Ensrud K, et al. (1998) Endogenous hormones and the risk of hip and vertebral fractures among older women. Study of Osteoporotic Fractures Research Group. N Engl J Med 339: 733-738. [Crossref]
- Christiansen M, Kveiborg M, Kassem M, Clark BF, Rattan SI (2000) CBFA1 and topoisomerase I mRNA levels decline during cellular aging of human trabecular osteoblasts. J Gerontol A Biol Sci Med Sci 55: B194-200. [Crossref]
- Nicolas V, Prewett A, Bettica P, Mohan S, Finkelman RD, et al. (1994) Age-related decreases in insulin-like growth factor-I and transforming growth factor-beta in femoral cortical bone from both men and women: implications for bone loss with aging. J Clin Endocrinol Metab 78: 1011-1016. [Crossref]
- Rosen CJ (1994) Growth hormone, insulin-like growth factors, and the senescent skeleton: Ponce de Leon's Fountain revisited? J Cell Biochem 56: 348-356. [Crossref]
- Sugimoto T, Nishiyama K, Kuribayashi F, Chihara K (1997) Serum levels of insulin-like growth factor (IGF) I, IGF-binding protein (IGFBP)-2, and IGFBP-3 in osteoporotic patients with and without spinal fractures. J Bone Miner Res 12: 1272-1279. [Crossref]
- Kodama Y, Takeuchi Y, Suzawa M, Fukumoto S, Murayama H, et al. (1998) Reduced expression of interleukin-11 in bone marrow stromal cells of senescence-accelerated mice (SAMP6): relationship to osteopenia with enhanced adipogenesis. J Bone Miner Res 13: 1370-7. [Crossref]
- Pfeilschifter J, Diel I, Scheppach B, Bretz A, Krempien R, et al. (1998) Concentration of transforming growth factor beta in human bone tissue: relationship to age, menopause, bone turnover, and bone volume. J Bone Miner Res 13: 716-30. [Crossref]
- Fleet JC, Cashman K, Cox K, Rosen V (1996) The effects of aging on the bone inductive activity of recombinant human bone morphogenetic protein-2. Endocrinology 137: 4605-4610. [Crossref]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, et al. (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390: 45-51. [Crossref]
- Kawaguchi H, Manabe N, Miyaura C, Chikuda H, Nakamura K, et al. (1999) Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J Clin Invest 104: 229-237. [Crossref]
- Manabe N, Kawaguchi H, Chikuda H, Miyaura C, Inada M, et al. (2001) Connection between B lymphocyte and osteoclast differentiation pathways. J Immunol 167: 2625-2631. [Crossref]
- Takahashi Y, Kuro-O M, Ishikawa F (2000) Aging mechanisms. Proc Natl Acad Sci U S A 97: 12407-12408. [Crossref]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, et al. (2005) Suppression of aging in mice by the hormone Klotho. Science 309: 1829-1833. [Crossref]
- Tatar M, Bartke A, Antebi A (2003) The endocrine regulation of aging by insulin-like signals. Science 299: 1346-1351. [Crossref]
- Kawano K, Ogata N, Chiano M, Molloy H, Kleyn P, et al. (2002) Klotho gene polymorphisms associated with bone density of aged postmenopausal women. J Bone Miner Res 17: 1744-1751. [Crossref]
- Ogata N, Matsumura Y, Shiraki M, Kawano K, Koshizuka Y, et al. (2002) Association of klotho gene polymorphism with bone density and spondylosis of the lumbar spine in postmenopausal women. Bone 31: 37-42. [Crossref]
- Yamada Y, Ando F, Niino N, Shimokata H (2005) Association of polymorphisms of the androgen receptor and klotho genes with bone mineral density in Japanese women. J Mol Med (Berl) 83: 50-57. [Crossref]
- Arking DE, Krebsova A, Macek M Sr, Macek M Jr, Arking A, et al. (2002) Association of human aging with a functional variant of klotho. Proc Natl Acad Sci U S A 99: 856-861. [Crossref]
- Arking DE, Becker DM, Yanek LR, Falli2021 Copyright OAT. All rights reservllele status and the risk of early-onset occult coronary artery disease. Am J Hum Genet 72: 1154-1161. [Crossref]
- Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC (2005) Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res 96: 412-8. [Crossref]
- Beresford JN (1989) Osteogenic stem cells and the stromal system of bone and marrow. Clin Orthop Relat Res 270-280. [Crossref]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, et al. (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284: 143-147. [Crossref]
- Bennett JH, Joyner CJ, Triffitt JT, Owen ME (1991) Adipocytic cells cultured from marrow have osteogenic potential. J Cell Sci 99: 131-139. [Crossref]
- Tontonoz P, Hu E, Spiegelman BM (1994) Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79: 1147-1156. [Crossref]
- Meunier P, Aaron J, Edouard C, Vignon G (1971) Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin Orthop Relat Res 80: 147-154. [Crossref]
- Burkhardt R, Kettner G, Bohm W, Schmidmeier M, Schlag R, et al. (1987) Changes in trabecular bone, hematopoiesis and bone marrow vessels in aplastic anemia, primary osteoporosis, and old age: a comparative histomorphometric study. Bone 8: 157-164. [Crossref]
- Rozman C, Feliu E, Berga L, Reverter JC, Climent C, et al. (1989) Age-related variations of fat tissue fraction in normal human bone marrow depend both on size and number of adipocytes: a stereological study. Exp Hematol 17: 34-37. [Crossref]
- Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung U, et al. (2004) PPAR insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest 113: 846-855. [Crossref]
- Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, et al. (1999) PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 4: 597-609. [Crossref]
- Kadowaki T (2000) Insights into insulin resistance and type 2 diabetes from knockout mouse models. J Clin Invest 106: 459-465. [Crossref]
- Ogawa S, Urano T, Hosoi T, Miyao M, Hoshino S, et al. (1999) Association of bone mineral density with a polymorphism of the peroxisome proliferator-activated receptor gamma gene: PPAR gamma expression in osteoblasts. Biochem Biophys Res Commun 260: 122-126. [Crossref]
- Koutnikova H, Cock TA, Watanabe M, Houten SM, Champy MF, et al. (2003) Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR gamma hypomorphic mice. Proc Natl Acad Sci U S A 100: 14457-14462. [Crossref]
- Cock TA, Back J, Elefteriou F, Karsenty G, Kastner P, et al. (2004) Enhanced bone formation in lipodystrophic PPAR gamma(hyp/hyp) mice relocates haematopoiesis to the spleen. EMBO Rep 5: 1007-1012. [Crossref]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, et al. (2000) Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100: 197-207. [Crossref]
- Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, et al. (2002) Leptin regulates bone formation via the sympathetic nervous system. Cell 111: 305-317. [Crossref]
- Berner HS, Lyngstadaas SP, Spahr A, Monjo M, Thommesen L, et al. (2004) Adiponectin and its receptors are expressed in bone-forming cells. Bone 35: 842-849. [Crossref]
- Lenchik L, Register TC, Hsu FC, Lohman K, Nicklas BJ, et al. (2003) Adiponectin as a novel determinant of bone mineral density and visceral fat. Bone 33: 646-651. [Crossref]
- Shinoda Y, Yamaguchi M, Ogata N, Akune T, Kubota N, et al. (2006) Regulation of bone formation by adiponectin through autocrine/paracrine and endocrine pathways. J Cell Biochem 99: 196-208. [Crossref]
- Kadowaki T, Tobe K, Honda-Yamamoto R, Tamemoto H, Kaburagi Y, et al. (1996) Signal transduction mechanism of insulin and insulin-like growth factor-1. Endocr J 43: S33-41. [Crossref]
- Ogata N, Chikazu D, Kubota N, Terauchi Y, Tobe K, et al. (2000) Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J Clin Invest 105: 935-943. [Crossref]
- Akune T, Ogata N, Hoshi K, Kubota N, Terauchi Y, et al. (2002) Insulin receptor substrate-2 maintains predominance of anabolic function over catabolic function of osteoblasts. J Cell Biol 159: 147-156. [Crossref]