Abstract
Retrospective analysis utilizing “next generation sequencing (NGS)” was done on cancer tissue harvested from 14 patients prior to receiving MLN8237, a novel Aurora Kinase A inhibitor. The responding patients (n=4) were characterized by stable disease ≥6 months and prolonged time of progression (≥1.3 fold prior treatment). Differential patterns of nodal connectivity in protein-protein interaction networks (consequent to determined genomic alterations) emerged from the comparison between responder and non-responder groups. The responding patient population showed high connectivity within MYC related genes including regulators of the Wnt/beta-catenin pathway. On the other hand, the non-responding patients showed high connectivity centered on the TP53/RB1 axis. Matching “targeted therapy to target” is a sine qua non for maximizing effective therapy in appropriate patients and NGS mapping may further our understanding of the relationships between molecular biological pathways and targeted therapy response. While awaiting further progress in systems analysis across “omic” levels (genomic-transcriptomic-proteomic), research involving of NGS sequence mapping to interrogate patient response to therapy in order to help elucidate molecular therapeutic predictors is justified based on the urgent needs of patient care.
Key words
aurora kinase a inhibitor, mln8237, wnt/beta-catenin , tp53/rb1 axis
Introduction
“Personalized” oncology, defined as the delivery of rationally based singlet or combinatorial therapeutics targeting a patient’s tumor-specific rewired pathway dysfunctional operational sites, has rapidly become the current paradigm of cancer treatment [1]. Despite consensus on this strategy, tactical implementation remains limited in scope [2]. The most appropriate methodology of target identification, including sequential parallel qualitative and quantitative retrieval of “omics” strata (i.e., genomics, epigenomics, transcriptomics, proteomics and metabolomics), data interrogation, and systems analysis has yet to be identified. However, the exigencies of patient care require the application of best available resources.
Of interest in this space is the regulation and targeting of Aurora kinase signaling. Aurora Kinase A (AURKA) is a highly conserved serine/threonine kinase [3], which is overexpressed or amplified in human cancer [4,5] and cancer cell lines [6-9]. Although AURKA is expressed in all actively dividing cells, overexpression is associated with oncogenesis. There is both cell cycle dependent (mRNA and protein elevation in G2-M followed by decrease in M-G1) and spatial modulation of AURKA [10]. It is localized to the centrosomes and the proximal mitotic spindles during mitosis where it functions in a diverse set of mitotic processes including centrosome maturation, bipolar spindle assembly, mitotic entry, chromosome alignment, and cytokinesis [10]. Ectopic expression of AURKA transforms rodent fibroblasts in culture and induces hyperplasia and mammary tumors when expressed in transgenic mice [11,12], which supports evidence of an oncogenic function of Aurora A in cancer. Elevated expression of AURKA has been shown to correlate with decreased survival in a variety of cancer types [13-17] and inhibition was associated with tumor regression in xenografted tumors [18]. The function of AURKA depends upon its ability to bind microtubules and localize to the centrosome and spindle poles where it phosphorylates and activates CDC25B phosphatase, which leads to activation and functional regulation of the Cyclin B/CDK1 complex [19,20]. AURKA also plays a role in the activation of PLK1, which contributes to both CDC25 and CDK1 activation [21-23].
While the interactions between p53 and AURKA are complex, there is emerging preclinical evidence that cancer cells lacking p53 function may be more resistant to Alisertib therapy. Specifically, a recent study demonstrated that triple negative breast cancer (TNBC) cells with loss of p53 function responded to Alisertib treatment by entering a state of cellular senesces, whereas p53-wt TNBC cells treated with Alisertib largely underwent apoptosis [24]. Furthermore, TNBC patient-derived xenograft models from patients who exhibited resistance to Alisertib showed a senescent (rather than an apoptotic) phenotype [24]. Another critical function of AURKA is stabilization of c-Myc, a well-characterized oncogene [25] which, in turn, upregulates AURKA.
Several inhibitors of AURKA/B including Hesperadin (B>A), MLN8237 (A>B), ZM447439 (B>A), VX680 (A=B) and AZD1152 (B>>>A) have been developed as anti-cancer agents with encouraging anti-tumoral potential in vitro and in vivo [18,26-31]. However, the underlying tumor-specific mechanisms of the anti-neoplastic activities of these drugs are still poorly understood.
We have evaluated DNA signal defects via available customized next generation sequencing (NGS) platforms in patients entered into a pilot trial (unpublished) with Alisertib (MLN8237), an adenosine triphosphate (ATP)-competitive/reversible inhibitor of both AURKA [28,29,31] and Aurora Kinase B (AURKB) [32,33], with greater AURKA specificity in vitro (A>B), to determine the translational potential of single level “omics” analysis in uncovering therapeutic predictive biomarkers. We performed a retrospective DNA molecular pathway analysis of cancer tissue from 14 consecutive responsive and non-responsive cancer patients entered into a prior unpublished pilot trial with Alisertib (MLN8237) which, although the ATP binding site affinity of AURKA for MLN8237 is higher than that of Aurora B, is likely inhibitory of both Aurora A and B kinases at therapeutic levels, at least in some tumors [33,34]. This preliminary analysis was intended to gain insight on the relationship of relevant molecular signals and cancer responsiveness to MLN8237.
Materials and methods
Patient population
Fourteen cancer patients (Demographics Table 1) were entered into study from 07/01/2013 to 04/01/2014. All patients received Alisertib(MLN8237) as part of participation study C14015 with Takeda (Cambridge, MA) (50mg BID for 7 days of a 21 day cycle starting at cycle 3; Cycle 1: 50mg QD on Day 1 and 10, 50mg BID Day 4-9 of a 21 day cycle; Cycle 2: 50mg QD on Day 8, 50mg BID on days 11-17 of a 28 day cycle) as part of a Phase I clinical trial MC #12-18. Patients were monitored for safety, response and survival as part of study MC #12-18. Archival tissue was retrospectively sent for molecular signal analysis to either Foundation Medicine (www.foundationmedicine.com) or Molecular Health (www.molecularhealth.com) for gene sequence analysis. All patients signed IRB approved consent for participation in study MC #12-18.
Table 1. Demographics of MLN8237 Treated Patients
|
Patient's Study ID
|
Cancer
|
Stage at Screening
|
Genome signals (amplified, mutated)
|
Therapy
(treatments prior to MLN8237)
|
Best Response/ Time to Progression
(prior tx)
|
|
501
|
Ovarian
|
III
|
AR, TP53, MCL1, NFKBIA a
|
Carbo + Taxol
Letrozole
Doxil
Topotecan
Carbo + Taxol + Custirsen
|
N/A
N/A
SD/ No PD
PD/ 84 days
SD/ 139 days
|
|
503
|
Ovarian
|
IV
|
ENG, pG191D b
|
Carbo + Taxol
Carboplatin + Taxol
Carboplatin + Gemcitabine
Gemcitabine alone
Doxil
|
N/A
CR/ 249 days
SD/ 241 days
SD/ 161 days
|
|
504
|
Breast
|
IV
|
AURKA, PTEN a
|
Carbo/ Taxol/Tamoxifen
Femara
Faslodex
Xeloda
Eribulin
Ixempra
Exemestane/Everolimus
Navelbine
Doxil
Cytoxan
|
N/A
N/A
N/A
SD/ 126 days
PD/ 70 days
PD/ 61 days
SD /131 days
PD/ 144 days
SD/ 96 days
PD/ 81 days
PD/ 15 days
|
|
505
|
Pancreatic
|
IV
|
PIK3CD, TSC1, STK11 b
|
Gemcitabine + Erlotinib
Capecitabine + Ruxolitinib
|
PD/ 60 days
SD/ 390 days
|
|
506
|
Liver
|
IV
|
Insufficient tissue
|
5-FU + Leucovorin
Carbo + Taxol
|
N/A
SD/ 99 days
|
|
507
|
Neuro-endocrine Carcinoma
|
IV
|
UNK
|
Carboplatin + VP-16
|
PD/98 days
|
|
508
|
Ovarian
|
IV
|
UNK
|
Carboplatin + Taxol (adjuvant)
Carboplatin + Taxol
Doxil
Topotecan
|
N/A
PD/ 133 days
PD/ 84 days
PD/ 76 days
|
|
509
|
Thymoma
|
IV
|
STK11 (LKB1) a
|
Cisplatin, Adriamycin, Cytoxan (adjuvant)
Cisplatin + XRT
|
N/A
PR/544 days
|
|
510
|
Colon
|
IV
|
APC, BRAF, KRAS, SMAD4, TP53 a
|
FOLFOX + Avastin (adjuvant)
FOLFIRI + Avastin (adjuvant)
5-FU + Leucovorin + Avastin + CPT 11
Imprime PGG + Erbitux
Xeloda + Perifosine
Investigational Agent (CDX 1127)
|
N/A
N/A
SD/258 days
PR/483 days
PR/507 days
PD/55 days
|
|
511
|
Ovarian
|
IV
|
MYC, CRKL, BRCA1 a
|
Carbo + Taxol
Doxil
Tamoxifen
Cisplatin + Gemzar
Gemzar - maintenance
Tamoxifen
Carboplatin
Taxol
Topotecan
Cistplatin + Gemzar
Carbo + Taxol + Custirsen
ONT-10
|
N/A
N/A
N/A
CR/ 954 days
CR cont’d/ 801 days
PD/ 102 days
PR/ 766 days
PD/ 126 days
SD/ 203 days
PR/ 175 days
PR/ No PD
SD/ 120 days
|
|
512
|
Pancreatic
|
IV
|
CCNE1, KRAS, RB1, TP53 a
|
Gemzar/ 5-FU + XRT (adjuvant)
FOLFIRINOX
Abraxane/Gemzar
|
N/A
SD/538 days
PD/62 days
|
|
513
|
Breast
|
IV
|
AURKA, PTEN, TP53 a
|
Adriamycin + Cytoxan
Tamoxifen
Taxotere
Doxil
Gemzar
Eribulin
Ixempra
Navelbine
SAR245409
|
N/A
N/A
CR/ 567 days
NE
PD/ 73 days
SD/ 123 days
PD/ unk
SD/ 349 days
SD/
|
|
514
|
NSCLC
|
IV
|
Insufficient tissue
|
Carboplatin + VP-16
Topotecan
|
CR/221 days
PD/ 95 days
|
|
515
|
Colon
|
IV
|
APC, KIT, TP53 a
|
FOLFOX
FOLFIRI
FOLFIRI + Vectibex
FOLFOX/Avastin
CEP-37250/KHK2804
|
N/A
PD/ 51 days
PR/ 407 days
SD/ 208 days
NE
|
|
aTargeted NGS methods
bWhole exome sequencing method
|
Gene sequencing
All Tumor tissue DNA extraction, library preparation and NGS analysis was performed by Foundation Medicine, Inc (Cambridge, Massachusetts), or Molecular Health (The Woodlands, Texas). A minimum of 50 ng of DNA extracted from pathologist-reviewed, Formalin-fixed, paraffin-embedded blocks. Tumor tissue was identified and micro-dissected, followed by DNA extraction and NGS library preparation consistent with established or proprietary methods. For Foundation analyzed samples, Genomic libraries were captured to analyze the whole exonic regions of 236 cancer-related genes and 47 introns of 19 genes commonly rearranged in cancer. For Molecular Health analyzed samples, genomic libraries were captured to analyze the whole exome or exonic regions of 617 genes; including oncogenes, tumor suppressors, other cancer-related genes, and genes of established pharmacogenomic importance. All samples were sequenced to high, uniform coverage (average of >95% of exons covered at greater than ×100), using Illumina HiSeq 2000 and Illumina HiSeq 2500 platforms. Genomic alterations (base substitutions, small insertions/deletions (INDELs), some rearrangements, and copy-number alterations) were determined. Potentially actionable alterations included those linked to anticancer drugs on the market or in registered clinical trials, excluding known benign SNPs (via dbSNP) and variants of unknown significance that were not predicted to influence gene function.
Protein-protein interaction networks
To generate networks, lists of genes affected by potentially actionable mutation were compiled from the lists of reported mutations in tables 1 and 2. For the non-responder patient population, all genes containing reported mutations were used. For the responder patient population, only genes that were uniquely mutated in this population (i.e., genes that were found to have reported mutations in the responder patients but not the non-responder patients) were used. These gene lists were analyzed using FunCoup v 3.0 build 2014-02 [35,36] a publically available, optimized Bayesian framework gene interaction analysis tool that can be found at: http://funcoup.sbc.su.se/search/. The analysis was restricted to protein-protein interactions (PPI) that have been annotated to occur within humans (interactions that have only been observed in non-human species were excluded). The resulting PPI networks were visualized using jsquid [37] a java-based application for the visualization and analysis of protein-protein interaction and functional coupling networks: http://jsquid.sbc.su.se/.
Results
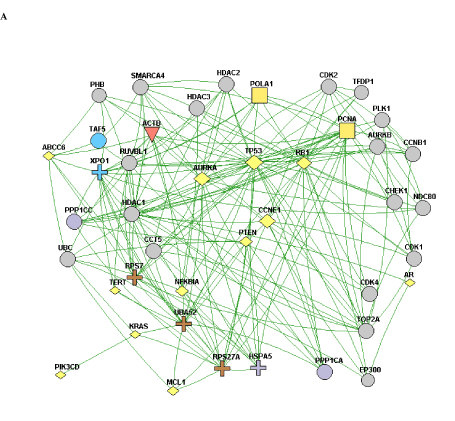
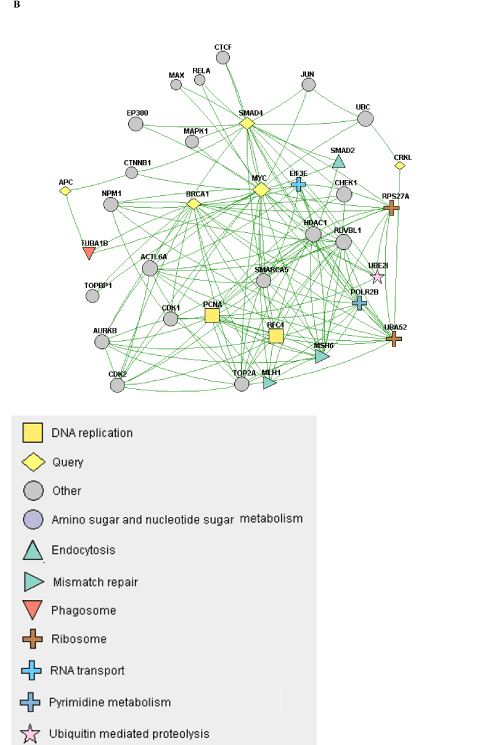
A summary of patient responses and survival to MLN8237 is shown in relation to molecular abnormalities in Table 2. Molecular profiling of cancer tissue was preferred independent and prior to entry into trial with Alisertib in patients with available paraffin stored tissue. Prolonged stable disease (SD) ³6 months from time of start of MLN8237 was observed in four patients, 506 (9 months), 509 (20 months), 510 (11 months), and 511 (14 months). Patients 509, 510 and 511 were evaluated by NGS methods. Patient 506 had insufficient tissue for molecular evaluation. An inactivating STK11 mutation was found in the thymoma of patient 509, an expected mutation of APC and a p53 mutation (R282W) of undefined functional significance in the colon cancer of patient 510, and c-Myc amplification (8 fold) in the ovarian cancer of patient 511. The genomic changes identified in patients 509, 510 and 511 share nodal connectivity to AURKA and AURKB, the expression of which or lack thereof has been shown to be complicit in cancer progression (i.e. most particularly vis-à-vis c-Myc expression with enhanced stabilization). Moreover, time of progression was significantly greater when comparing the time to progression on Alisertib to that achieved with the immediate prior cancer treatment for two of these patients (363 versus 55 days in 510, 426 versus 120 days in 511). All 3 of these patients also remain alive well beyond 1 year after treatment initiation (Table 2). The ten other patients (501, 503, 504, 505, 507, 508, 512, 513, 514 and 515) did not achieve SD ³6 months or better and none experienced a delay >1.3-fold in time to progression longer than their prior treatment. Von Hoff and colleagues [38] have suggested a ratio of >1.3 of time to progression with new therapy vs. time to progression with prior therapy as a surrogate measure of positive response to the new therapy). NGS was performed in five of these patients (504, 513, 501, 512, 515); two (503, 505) utilized whole exome sequencing and in three sequencing was not done (506, 507, 508). No actionable mutations were obtained for 515.
Table 2. Summary of Patient Responses and Survival to MLN8237 Shown in Relation to Molecular Abnormalities
|
Patient's Study ID
|
# of Cycles
|
Reason for Ending Treatment
|
Best Response 2 Months
|
Survival from start of MLN8237 therapy (days)
|
Response time from start of MLN8237 therapy
|
Validated cancer-associated
mutations
|
|
501
|
2
|
Disease Progression
|
PD
|
240
|
45
|
TP53.pA276fs69, AR.pD840N,
NFKBIA amp,
MCL1 amp
|
|
503
|
2
|
Disease Progression
|
PD
|
206
|
60
|
ENG.pG191D, TP53.pR249S,
TERT.pH412Y
|
|
504
|
2
|
Disease Progression
|
PD
|
153
|
51
|
PTEN.pI101fs12, AURKA amp
|
|
505
|
2
|
Disease Progression
|
PD
|
83
|
48
|
STK11.pQ100, PIK3CD.pS520A,
ABCC6.pR265G
|
|
506
|
12
|
Disease Progression
|
SD
|
625
|
272
|
N/A
|
|
507
|
2
|
Clinical Progression
|
SD
|
76
|
76
|
N/A
|
|
508
|
4
|
Disease Progression
|
SD
|
270
|
98
|
N/A
|
|
509
|
17
|
**
|
SD
|
429
|
369
|
STK11.pF354L
|
|
510
|
11
|
Toxicity
|
SD
|
423
|
363
|
KRAS.pG12S, APC.pD1569fs74,
TP53.pR282W, APC.pR213, SMAD4.pQ516
|
|
511
|
14
|
**
|
SD
|
409
|
344
|
BRCA1.pE1046, CRKL amp, MYC amp
|
|
512
|
<2
|
Clinical Progression
|
NE
|
22
|
UNK
|
KRAS.pG12V, TP53.pG245S, RB1.pY454,
CCNE1 amp
|
|
513
|
2
|
Disease Progression
|
PD
|
168
|
55
|
PTEN.pF238fs20, TP53.pV225fs17,AURKA amp
|
|
514
|
<2
|
Disease Progression
|
PD
|
73
|
19
|
N/A
|
|
515
|
2
|
Disease Progression
|
PD
|
516
|
49
|
APC, KIT, TP53
|
|
** Indicates patient is currently receiving treatment with MLN8237 (Alisertib)
Updated data as of 11/07/2014
|
2021 Copyright OAT. All rights reserv
When the constellation of genes that are mutated/copy-altered in the responder and non-responder groups are analyzed, differential patterns of nodal connectivity in protein-protein interaction networks emerge (Figure 1). A network developed from the genes that are mutated/copy-altered in the non-responder patient population is centered on the TP53/RB1 axis, with extensive connectivity between TP53 and a number of genes that were mutated/amplified in the non-responding patient population. This is consistent with preclinical findings which suggest that loss of p53 function may promote resistance to Alisertib [24]. While there was a single TP53 mutation (R282W) identified in the responder patient 510, there is some structural evidence that this particular p53 mutation may retain some functionality [39]. Interestingly, a network formed from genes that were uniquely mutated/copy-altered in the responding patient population is centered on MYC and MYC-related genes, including negative regulators of the Wnt/beta-catenin pathway. This is particularly intriguing considering the established pattern of cross-regulation between MYC and the Wnt/beta-catenin pathway [40,41].
Discussion
Alisertib is being developed as a small molecular inhibitor of AURKA (although, as noted above, it is likely differentially inhibitory of both Aurora A and B kinases at therapeutic levels) for the treatment of advanced malignancies and has already demonstrated activity against a broad range of both in vitro and in vivo preclinical tumor models.
NGS based evaluation of the cancer genome with consequent protein-protein interaction mapping, although a first step in tumor biomolecular deconstruction, is a key component in the personalization of cancer therapy. Knowledge of the mutated genes and variants of the responsive and non-responsive populations resulted in construction of two different gene-gene protein interaction networks; one (Figure 1B) representing the non-responsive patients and one (Figure 1A) the responsive patients. As such two distinctly different networks were constructed based on the different gene mutation profile. This analysis was performed to see if there was a specific pathway/interaction cluster that was uniquely mutated in the responder population but not the non-responder population and in doing so provide suggestive direction in interpreting relationship of said pathway to predictive opportunity for response and to determine relationship of said pathway to mechanism of Alisertib. In our pathway network assessment the Wnt/beta-catenin pathway appeared to be mutated exclusively in the responder population, while other “non” Wnt/beta-catenin pathways were identified in the non-responder group. Based on the very small number of assessable patients, results can only be considered as suggestive and hypothesis generating. Statistical significance was not achieved. The approach in this study has highlighted a possible relationship between MYC expression and sensitivity to Alisertib. That AURKA has a critical function in stabilizing N-Myc protein was initially reported by Otto et al. [25] in neuroblastoma, half of which carry N-Myc amplification. They showed that elevated levels of AURKA inhibit the degradation of Myc during mitosis by interacting with both Myc and the Fbxw7 ubiquitin ligase. As a result, high expression levels of AURKA effectively uncouple degradation of Myc from PI3-kinase-dependent signaling. Interestingly, our cohort showed a dearth of PI3K/AKT/mTOR mutations in the responder group, consistent with the above observation. This may be because MYC-dependence on the PI3K/AKT/mTOR pathway is relieved by a positive feedback loop involving either MYC-AURKA or MYC-Wnt/beta-catenin. Aberrant activation of the Wnt/beta-catenin signaling pathway is associated with numerous cancers and indeed correlates frequently with amplification of c-Myc oncogene or c-Myc related signaling. Co-expression of c-Myc and Wnt-1 in nude murine models is associated with rapid tumor growth. It appears that the anti-apoptotic function of Wnt-1 plays a critical role in synergistic action between c-Myc and Wnt-1 [42]. Our findings suggest that anything that promotes MYC (including just direct MYC amplification) in the absence of AKT/PI3K/mTOR elements may predict sensitivity to ALS. Furthermore, Myc both directly and indirectly upregulates AURKA transcription, a process essential for the maintenance of the malignant state [43]. c-Myc destabilization is one of the mechanisms resulting in anticancer activity demonstrated in tumor xenografts with Alisertib [44]. Notably, the ovarian cancer from patient 511 demonstrated c-Myc amplification. That no relationship was shown towards sensitivity of the two AURKA amplified patients to Aurora Kinase inhibitor highlights the need for eventual full “omics” integration and multilevel systems analysis.
Figure 1. A) A protein-protein interaction network of genes with reported variants in the non-responder patient population. B) A protein-protein interaction network of genes with reported variants unique to the responder patient population.
A
B
In the three “responders” with molecular characterization, there appears to be an overrepresentation of mutations in the Wnt/β-catenin and TGFβ pathways in the patients who exhibited the best response to Alisertib therapy. This overrepresentation was detected by gene ontology clustering analysis, although it fails to reach statistical significance, most likely due to the small sample size. The colorectal adenocarcinoma carcinoma of patient 510 is characterized by multiple genetic changes involving APC, KRAS, BRAF, TP53 and SMAD4. As in patient 511, Myc again emerges as a likely common nodal target interactive with AURKA. SMAD4 and AURKA interact via a reciprocal TGFβ-independent pathway and the former blocks the direct and indirect up-regulation of Myc (AURKA inhibits GSK3β which in turn inhibits β-catenin/TCF which regulates Myc). The mutated SMAD4 would, then, effectively increase Myc expression. In addition, insofar as LKB1 (STK11) interacts with APC to downregulate Wnt/TCF and Myc, the loss of APC would, likewise, result in upregulation of Myc expression [45,46]. There are multiple levels of interaction between the Myc and Wnt/TCF pathways that are observed in these patients. Furthermore, overexpression of Myc, loss of negative regulators of Wnt/beta-catenin signaling (such as SMAD4 and APC), as well as damaging mutations in TGF-beta may be predictive of cells that are AURKA-driven and thus sensitive to Alisertib [47].
Using in vitro signaling studies, Alisertib has been shown to lead to G2/M arrest in both breast (MCF7 (p53 wt) and MDA-MB 231 (p53 C839G>A) [48] and osteosarcoma (U-2 OS (p53 wt) and MG-63 (p53 wt) [49] cell models, in both cases via activation of pro-apoptotic signaling (lowered BCL-2, upregulated Bax) and downregulation of PI3K/AKT/mTOR signaling. That this mechanism may be partially p53-dependent is supported by a recent study that demonstrated that triple negative breast cancer (TNBC) cells with loss of p53 function responded to Alisertib by entering cellular senescence, whereas p53-wt TNBC cells underwent apoptosis [24]. Furthermore, TNBC-PDX models from patients with resistance to Alisertib show a senescent phenotype [24]. Although one responding patient (PID510) had a TP53 mutation (R282W) it is notable that this mutation remains of undefined functional significance [39,50,51]. In a reciprocal negative feedback interaction, wild-type p53 is regulated by AURKA phosphorylation, which, in turn, inhibits interaction with MDM2 [52] and p53 functions as a negative regulator of AURKA via both transcriptional and translational modifications [53]. Therefore, a loss-of-function mutation in TP53 could result in enhanced expression of AURKA and increased sensitivity to targeted inhibitory therapy [53-55]. Yet, two recent studies show that MK-8745 (A>>B) can utilize both p53-dependent [56] and p53-independent [57] mechanisms. Whether this apparent mechanistic disparity is due to microenvironmental differences, differences in G2-M slippage due to kinetics of the cyclin B1 protein [58] or susceptibility to p53 mediated G1 checkpoint arrest remains undetermined. Alternatively, insofar as AURKB inhibition bypasses the G2-M checkpoint and thereby activates G1 checkpoint activity resulting in apoptosis due to the accumulation of chromosomal instability, it is possible that Aliserib may act as a pan-AURK inhibitor in a dose-dependent/tumor-dependent manner [33,34,58]. Thus, although brought to attention as a potential indicator of responsiveness by NGS analysis, the role of p53, which appears to be contextual, remains to be further elucidated.
As this very preliminary evaluation shows, despite the implementation of NGS as an adjunct to optimizing the choice of personalized therapeutics, obvious limitations are evident. This tool is currently unable to document DNAàRNA sequence discordance or RNAàprotein expression discordance [59,60]. For example, two patients (504 and 513) with AURKA amplification did not respond to Alisertib. Insofar as AURKA copy numberàprotein expression discordance has been described [16], without stratified “omic” assessment the reason for lack of response cannot be ascertained, e.g., whether lack of AURKA protein overexpression, discordance between protein abundance and basal phosphorylation [61], or pathway signaling dependence on dynamic quantitative versus qualitative protein expression levels [62].
Matching targeted therapy to target, including multiple target enumeration based on pathway crosstalk and feedback, is a complicated process that requires further discovery. However, while our multi-strata “omic” toolbox continues to expand its capabilities, patients are in need of care. Although only a first step, the application of NGS to target assessment has now become patient-ready and can supplement existing tools to provide further increments in treatment outcome.
Acknowledgements
We gratefully acknowledge the generous support of Michele Ashby, as well as the MMK Foundation.
References
- Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, et al. (2011) Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med 3: 111ra121. [Crossref]
- Nemunaitis J (2014) Genome Expression to Targeted Therapy in Cancer Management. Hereditary Genet 3: 129.
- Vas AC, Clarke DJ (2008) Aurora B kinases restrict chromosome decondensation to telophase of mitosis. Cell Cycle 7: 293-296. [Crossref]
- Fu J, Bian M, Jiang Q, Zhang C (2007) Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 5: 1-10. [Crossref]
- Keen N, Taylor S (2004) Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 4: 927-936. [Crossref]
- Katayama H, Ota T, Jisaki F, Ueda Y, Tanaka T, et al. (1999) Mitotic kinase expression and colorectal cancer progression. J Natl Cancer Inst 91: 1160-1162. [Crossref]
- Sasai K, Katayama H, Stenoien DL, Fujii S, Honda R, et al. (2004) Aurora-C kinase is a novel chromosomal passenger protein that can complement Aurora-B kinase function in mitotic cells. Cell Motil Cytoskeleton 59: 249-263. [Crossref]
- Tatsuka M, Katayama H, Ota T, Tanaka T, Odashima S, et al. (1998) Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res 58: 4811-6. [Crossref]
- Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, et al. (1998) Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 20: 189-193. [Crossref]
- Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA (2013) Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci 70: 661-687. [Crossref]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, et al. (1998) A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 17: 3052-3065. [Crossref]
- Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, et al. (2006) Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 25: 7148-7158. [Crossref]
- Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, et al. (2008) Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res 14: 4455-4462. [Crossref]
- Goos JA, Coupe VM, Diosdado B, Delis-Van Diemen PM, Karga C, et al. (2013) Aurora kinase A (AURKA) expression in colorectal cancer liver metastasis is associated with poor prognosis. Br J Cancer 109: 2445-2452.[Crossref]
- Xu J, Wu X, Zhou WH, Liu AW, Wu JB, et al. (2013) Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS One 8: e56919. [Crossref]
- Yamamoto S, Yamamoto-Ibusuki M, Yamamoto Y, Fujiwara S, Iwase H (2013) A comprehensive analysis of Aurora A; transcript levels are the most reliable in association with proliferation and prognosis in breast cancer. BMC Cancer 13: 217. [Crossref]
- Zeng B1, Lei Y1, Zhu H1, Luo S1, Zhuang M1, et al. (2014) Aurora-A is a novel predictor of poor prognosis in patients with resected lung adenocarcinoma. Chin J Cancer Res 26: 166-173. [Crossref]
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, et al. (2004) VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 10: 262-7. [Crossref]
- Cazales M, Schmitt E, Montembault E, Dozier C, Prigent C, et al. (2005) CDC25B phosphorylation by Aurora-A occurs at the G2/M transition and is inhibited by DNA damage. Cell Cycle 4: 1233-1238. [Crossref]
- Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, et al. (2004) Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci 117: 2523-2531.[Crossref]
- Macůrek L1, Lindqvist A, Lim D, Lampson MA, Klompmaker R, et al. (2008) Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 455: 119-123.[Crossref]
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G (2008) Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 320: 1655-1658.[Crossref]
- Yuan K, Huang Y, Yao X (2011) Illumination of mitotic orchestra during cell division: a Polo view. Cell Signal 23: 1-5. [Crossref]
- Tentler JJ, Ionkina AA, Tan AC, Newton TP, Pitts TM, et al. (2015) p53 Family Members Regulate Phenotypic Response to Aurora Kinase A Inhibition in Triple-Negative Breast Cancer. Mol Cancer Ther 14: 1117-29. [Crossref]
- Otto T1, Horn S, Brockmann M, Eilers U, Schüttrumpf L, et al. (2009) Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15: 67-78. [Crossref]
- Lee EC1, Frolov A, Li R, Ayala G, Greenberg NM (2006) Targeting Aurora kinases for the treatment of prostate cancer. Cancer Res 66: 4996-5002. [Crossref]
- Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, et al. (2007) AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res 13: 3682-3688. [Crossref]
- Venkatakrishnan K, Zhou X, Ecsedy J, Mould DR, Liu H, et al. (2014) Dose Selection for the Investigational Anticancer Agent Alisertib (MLN8237): Pharmacokinetics, Pharmacodynamics, and Exposure-Safety Relationships. J Clin Pharmacol [Crossref]
- Goldberg SL, Fenaux P, Craig MD, Gyan E, Lister J, et al. (2014) An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leukemia Research Reports 3: 58-61.
- Malumbres M, Pérez de Castro I (2014) Aurora kinase A inhibitors: promising agents in antitumoral therapy. Expert Opin Ther Targets 18: 1377-1393. [Crossref]
- Falchook G, Kurzrock R, Gouw L, Hong D, McGregor KA, et al. (2014) Investigational Aurora A kinase inhibitor alisertib (MLN8237) as an enteric-coated tablet formulation in non-hematologic malignancies: phase 1 dose-escalation study. Invest New Drugs 32: 1181-1187. [Crossref]
- Pinel S, Barbault-Foucher S, Lott-Desroches MC, Astier A (2009) Inhibitors of aurora kinases. Ann Pharm Fr 67: 69-77. [Crossref]
- Qi W, Spier C, Liu X, Agarwal A, Cooke LS, et al. (2013) Alisertib (MLN8237) an investigational agent suppresses Aurora A and B activity, inhibits proliferation, promotes endo-reduplication and induces apoptosis in T-NHL cell lines supporting its importance in PTCL treatment. Leuk Res 37: 434-439. [Crossref]
- Qi W, Cooke LS, Liu X, Rimsza L, Roe DJ, et al. (2011) Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol 81: 881-890. [Crossref]
- Schmitt T, Ogris C, Sonnhammer EL (2014) FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res 42: D380-388. [Crossref]
- Alexeyenko A, Sonnhammer EL (2009) Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res 19: 1107-1116. [Crossref]
- Klammer M, Roopra S, Sonnhammer EL (2008) jSquid: a Java applet for graphical on-line network exploration. Bioinformatics 24: 1467-1468. [Crossref]
- Von Hoff DD, Stephenson JJ Jr, Rosen P, Loesch DM, Borad MJ, et al. (2010) Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 28: 4877-4883. [Crossref]
- Calhoun S, Daggett V (2011) Structural effects of the L145Q, V157F, and R282W cancer-associated mutations in the p53 DNA-binding core domain. Biochemistry 50: 5345-5353. [Crossref]
- Xu J, Chen Y, Huo D, Khramtsov A, Khramtsova G, et al. (2015) β-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol Carcinog [Crossref]
- Karim R, Tse G, Putti T, Scolyer R, Lee S (2004) The significance of the Wnt pathway in the pathology of human cancers. Pathology 36: 120-128. [Crossref]
- You Z, Saims D, Chen S, Zhang Z, Guttridge DC, et al. (2002) Wnt signaling promotes oncogenic transformation by inhibiting c-Myc-induced apoptosis. J Cell Biol 157: 429-440. [Crossref]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, et al. (2010) Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 116: 1498-1505. [Crossref]
- Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, et al. (2011) Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol 68: 1291-304. [Crossref]
- Jian SF, Hsiao CC, Chen SY, Weng CC, Kuo TL, et al. (2014) Utilization of liquid chromatography mass spectrometry analyses to identify LKB1-APC interaction in modulating Wnt/β-catenin pathway of lung cancer cells. Mol Cancer Res 12: 622-635. [Crossref]
- Tsai LH, Wu JY, Cheng YW, Chen CY, Sheu GT, et al. (2015) The MZF1/c-MYC axis mediates lung adenocarcinoma progression caused by wild-type lkb1 loss. Oncogene 34: 1641-1649. [Crossref]
- Yochum GS, Sherrick CM, Macpartlin M, Goodman RH (2010) A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5' and 3' Wnt responsive enhancers. Proc Natl Acad Sci U S A 107: 145-150. [Crossref]
- Li JP, Yang YX2, Liu QL3, Pan ST4, He ZX5, et al. (2015) The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des Devel Ther 9: 1627-1652. [Crossref]
- Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH, et al. (2015) Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther 9: 1555-1584. [Crossref]
- Walker DR, Bond JP, Tarone RE, Harris CC, Makalowski W, et al. (1999) Evolutionary conservation and somatic mutation hotspot maps of p53: correlation with p53 protein structural and functional features. Oncogene 18: 211-218. [Crossref]
- Bullock AN, Henckel J, Fersht AR (2000) Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19: 1245-1256. [Crossref]
- Hsueh KW, Fu SL, Chang CB, Chang YL, Lin CH (2013) A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochim Biophys Acta 1834: 508-515. [Crossref]
- Wu CC, Yang TY, Yu CT, Phan L, Ivan C, et al. (2012) p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 11: 3433-3442. [Crossref]
- Kalous O, Conklin D, Desai AJ, Dering J, Goldstein J, et al. (2013) AMG 900, pan-Aurora kinase inhibitor, preferentially inhibits the proliferation of breast cancer cell lines with dysfunctional p53. Breast Cancer Res Treat 141: 397-408. [Crossref]
- Marxer M, Ma HT, Man WY, Poon RY (2014) p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases. Oncogene 33: 3550-3560. [Crossref]
- Nair JS, Ho AL, Schwartz GK (2012) The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle 11: 807-817. [Crossref]
- Shionome Y, Yan L, Liu S, Saeki T, Ouchi T (2013) Integrity of p53 associated pathways determines induction of apoptosis of tumor cells resistant to Aurora-A kinase inhibitors. PLoS One 8: e55457. [Crossref]
- Hilton JF, Shapiro GI (2014) Aurora kinase inhibition as an anticancer strategy. J Clin Oncol 32: 57-59. [Crossref]
- Chen G, Gharib TG, Huang CC, Taylor JM, Misek DE, et al. (2002) Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics 1: 304-313. [Crossref]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, et al. (2011) Widespread RNA and DNA sequence differences in the human transcriptome. Science 333: 53-58. [Crossref]
- Niepel M, Hafner M, Pace EA, Chung M, Chai DH, Zhou L, et al. (2013) Profiles of Basal and stimulated receptor signaling networks predict drug response in breast cancer lines. Sci Signal 6: ra84. [Crossref]
- Cohen-Saidon C, Cohen AA, Sigal A, Liron Y, Alon U (2009) Dynamics and variability of ERK2 response to EGF in individual living cells. Mol Cell 36: 885-893. [Crossref]