Abstract
Infectious erysipelas is a common clinical diagnosis in daily practice that usually responds well to standard antibiotic treatment. In cases of non-responders to conventional anti-infective therapies, two things need to be done: antibiotic resistance must be ruled and diagnosis has to be re-evaluated. Familial Mediterranean Fever (FMF) is the most common entity within the huge group of hereditary periodic fever syndromes. FMF is characterized by distinct clinical findings, especially the late clinical onset post-infancy and pathognomonic erysipelas-like eruptions. Against the background of autoinflammatory syndromes that may present with erysipelas-like eruptions, patient’s history has to be recorded very carefully and clinical and dermatopathologic findings have to be evaluated meticulously.
Key words
familial mediterranean fever, erysipelas, neutrophilic urticarial dermatosis, erysipelas-like eruption, hereditary periodic fever syndromes
Case report
A 55-year old female patient presented with a sharply circumscribed erythema of the gluteal area, suggestive for infectious erysipelas (Figure 1). She reported similar episodes of these eruptions in the recent years that had always been treated with oral antibiotics. Family and personal history were inconspicuous, especially for diabetes mellitus. Skin lesions were always conducted by pyrexia peaking at 40.5°C, fatigue, muscle, joint, and even chest pain, lasting for several days even though oral antibiotic treatment had been performed. In the past, these general symptoms were considered as being para-infectious defining bacterial erysipelas. Intriguingly, after termination of antibiotic therapy, the skin lesion locally recurred usually within a week presenting with the same clinical signs and symptoms. During the course of the disease different antibiotics had been unsuccessfully administered: penicillin, clindamycin and others. After ten months of recurring episodes of erythema and general infectious symptoms as reported above, the patient was admitted to our dermatology department for intravenous treatment with clindamycin. Laboratory investigations were normal and showed only insignificantly elevated ANAs. Rheumatoid factors, ferritin, angiotensin-converting enzyme (ACE) and lymphocytic subpopulation were within normal ranges. Merely, C-reactive protein and peripheral neutrophils were slightly elevated during the phases of clinically obvious inflammation.
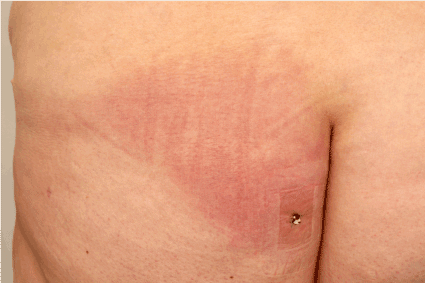
Figure 1. Clinical presentation with sharply demarcated erythema of the buttock.
Figure 2a. HE stain. Magnification 20x. Note dense perivascular and interstitial inflammatory infiltrate composed of lymphocytes and neutrophils devoid of epidermal changes or vascular damage.
Clinical signs and symptoms showed an undulating course, with phases of recovery and direct aggravation always being attended by episodes of high fever that did not fit to the clinical presentation of fading erythema. The case had been discussed several times with colleagues from the department of microbiology and finally antibiotic treatment was changed to the administration of linezolid for more than 28 days. Intriguingly, even the long-time administration of this last resort antibiotic did not lead to a clinical improvement.
Intensive investigations performed to rule out differential diagnoses (i.e., the adult form of Morbus Still or systemic vasculitis) did not show any abnormalities: there were no signs of organ involvement; liver enzymes were normal, urine analysis showed no proteinuria, and echocardiography did not reveal any signs of endocarditis, neither did abdominal sonography. Due to the gluteal location of the cutaneous erythema, the patient was examined proctologically and a magnetic resonance image (MRI) of the pelvis and chest was conducted that both showed no signs of vasculitis or hidden inflammatory foci. Hence, a skin biopsy was taken from lesional skin that revealed a regular epidermis, with a dense perivascular and interstitial infiltration of lymphocytes and lots of neutrophils (Figure 2a), but without nuclear debris or nuclear dust and without leukocytoclasia. Signs of vasculitis, e.g. destruction of vascular walls, transmural inflammatory cells or obstruction of blood vessels were not noted. There was no papillary edema and no evidence for bacteria or fungi in gram and PAS staining. Intriguingly, neutrophilic epitheliotropism of eccrine gland epithelium could be found (Figure 2b and c). Thus, erysipelas-like-eruption was diagnosed. The unsatisfying course of disease in our case prompted us to reconsider our diagnosis of erysipelas and intensive anamnesis of the patient revealed additional interesting information: our patient was of Turkish descent, her father was a Turk.
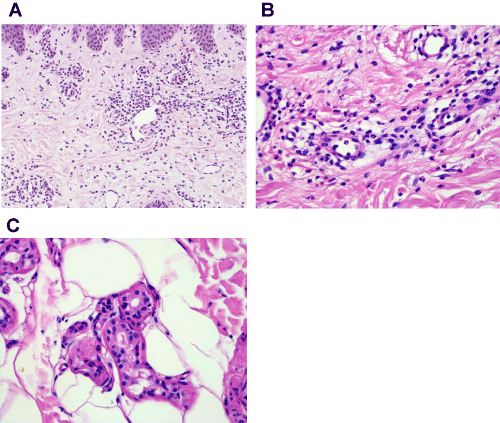
Figure 2b. HE stain. Magnification 40x. Please note the intact vascular wall and missing nuclear dust.
Figure 2c. HE stain. Magnification 40x. Neutrophils within secretory epithelia of sweat glands.
In summary our patient presented with an erysipelas-like-eruption refractory to antibiotic treatment accompanied by episodes of pyrexia and muscle-/ joint- and chest pain. Additionally our patient came from a Turkish family. Thus we finally changed the clinical diagnoses to Familial Mediterranean Fever and initiated a systemic therapy with colchicine. Only a few days later we observed paling of the erythema, disappearance of pyrexia and remission of general symptoms like muscle and joint pain. Now, several months later, the patient continues to be in full remission. Unfortunately, the patient did not give consent for further genetic analyses.
Introduction to autoinflammation
Autoinflammatory syndromes are rare, usually monogenetic diseases that represent challenging diagnoses for clinicians in general and dermatopathologists in particular. Uniformly, the term “hereditary periodic fever syndromes” (HPFS) is used to summarize distinct syndromes [1]. Due to clinical and morphological findings that may be interpreted as nonspecific, HPFS are often diagnosed with delay.
According to recent classification HPFS comprise four distinct entities: Familian Mediterranean Fever (FMF), cryopyrin-associated periodic syndrome (CAPS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS) and mevalonate kinase deficiency (syn. hypergammaglobulinaemia D) [1]. For comprehensive review on TRAPS see Schmaltz and colleagues [2]. Within the group of CAPS, familial cold urticarial syndrome (FCAS), Muckle-Wells syndrome (MWS) and chronic infantile neurological cutaneous and articular syndrome (CINCA) are subsumed [3].
Clinically these diseases are characterized by recurrent fever attacks and a variety of localized inflammation leading to symptoms in distinct organs, such as skin, serosal membranes, joints, central nervous system etc. [3]. Typically, most of these HPFSs manifest within the first year of life and for CAPS appearance of symptoms in early-childhood is usually observed.
Late disease onset has not yet been described in FMF as seen in our case and TRAPS. Genetic background are mutations in the MEFV gene for FMF and TNFRSF1A gene for TRAPS [3]. In contrast to these monogenic HPFSs polygenic entities are known, named NOD2-associated autoinflammatory diseases (AID), also appearing in adult age [4]. NOD (nucleotide-binding oligomerization domain containing 2) belongs to the family of intracellular proteins with N-terminal caspase recruitment domains and are associated with increased inflammatory response and enhanced autophagic activity [5].
FMF is the most common form of HPFS, it is characterized by an autosomal recessive inheritance and caused by a loss-of-function mutation in the MEFV gene typically affecting adults of the peri-Mediterranean area [1]. The MEFV gene encodes for a protein named Pyrin that is found in myeloid cells and thought to modulate cell susceptibility to apoptosis via regulation of IL-1β [1,6]. The MEFV gene is expressed in neutrophils, eosinophils, monocytes, dendritic cells and synovial fibroblasts [7]. Intriguingly, there is no evidence for functional differences between neutrophils from families with and without FMF, there are no morphologic changes between mutation carriers and healthy people and there are no changes in infection rates in patients with FMF [6]. There is rising evidence that mutations in the MEFV gene may lead to an altered regulation of inflammatory phagocyte-mediated response resulting in the distinct clinical phenotype of FMF [6]. Cutaneous involvement in FMF is rarely, but nevertheless typically and comprises recurrent erysipelas-like rashes of the inferior limbs as well as a variety of unspecific cutaneous symptoms such as diffuse erythema of the face and /or trunk, angioneurotic edema, pyoderma and subcutaneous nodules [1,8]. Erysipelas-like rashes are mainly tender, erythematous plaques over joints, legs and dorsal feet [8].
There is a distinction in phenotype I, that reveals the classical clinical phenotype and a genetically confirmation of mutation in the MEFV gene, a phenotype II, these patients develop a systemic amyloidosis without previous typical signs of FMF. A third phenotype III can be observed: these patients do not have any clinical signs but exhibit the mutation in MEFV gene [1]. Clinically, major and minor criteria for the diagnosis of FMF have been defined by different authors. The widely used Tel Hashomer criteria arrogate the presence of two major or one major and two minor criteria for establishing the diagnosis [9]. Following the Tel Hashomer criteria these major criteria include recurrent febrile episodes accompanied by serositis, presence of systemic amyloidosis or favorable response to colchicine. Minor criteria encompass recurrent febrile episodes, erysipelas- like skin rash and FMF in a first-degree relative. Genetic investigations with proof of mutation are able to confirm the diagnosis [1,9]. Aside this, the so called Livneh criteria and Yalcinkaya-Ozen criteria do exist with distinct sensitivity rates in different cohorts and age groups investigated [10,11]. Thus, in order for choosing the correct classification system, the clinician has to take the age of the patients into account. For a comprehensive review on the different classification systems see P. Portincasa [7].
Clinical course and treatment
In the case presented herein, concerning to the Tel Hashomer system our patient fulfills two major criteria (recurrent febrile episodes with serositis {joint pain as sign for synovial serositis and chest pain as a symptom of pleuritis} and a favorable response to colchicine) and one minor criterion (erysipelas –like skin rashes). Thus, the diagnosis of FMF was established. Clinical differential diagnoses encompass erysipelas, erysipelas-like rashes after joint replacements, mercury exanthemas and malignant counter-parts (e.g. erysipelas melanomatosa) respectively paraneoplastic rashes [12-14]. The lack of response to suitable antibiotics in adequate dosage also prompted us towards a non- infectious origin of the recurrent erythema. General symptoms that were considered as parainfectious appeared sternly rhythmical with and without antibiotic treatment what also supports the hypothesis of a non-infectious origin. Histomorphology of a skin biopsy prompted us to perform an extensive work-up to rule out differential diagnoses of autoinflammatory syndromes, especially erysipelas-like rashes in the course of FMF.
Treatment of FMF has to be initiated as soon as possible to prevent acute febrile painful attacks and especially the development of a systemic AA-amyloidosis. First-line treatment of FMF is colchicine [7]. Alternatively, in cases of intolerance or non-responders TNFα-inhibitors (e.g. etanercept), IL-1 inhibitors (e.g. anakinra) and IL-6 receptor antibodies (e.g. tocilizumab) may be used [7]. Since 1972 the successful use of colchicine in FMF is documented [15]. Though, patients have to be monitored under therapy with colchicine, administration of colchicine is recommended to be continued life-long due to early relapses and hazard of FMF-related complications [7]. Distinct recommendations concerning therapy of FMF, especially in colchicine intolerance or non-responders are given by Portincasa P [7].
Dermatopathologic findings in HPFS
The main challenge for dermatopathologists faced with autoinflammatory febrile diseases is to think of it as a differential diagnoses with minimal specific morphologic changes in punch biopsies especially in the context of missing clinical information. Erysipelas-like rashes are within the spectrum of neutrophilic dermatoses with distinct and even unobtrusive morphologic changes. Histopathologically, slight papillary edema can be observed, discrete superficial and deep lymphocytic infiltration with some admixed neutrophils. Eosinophils are mostly absent. Usually no vasculitis-typical changes are seen (missing transmural infiltration, absence of nuclear dust and no extravasation of erythrocytes)[16]. However, one report of leucocytoclastic vasculitis in a case of erysipelas-like rash does exist [8]. Torrelo and colleagues presented a case series of six patients with CANDLE syndrome (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature) and did a comprehensive immunophenotypical work-up on these biopsy specimens [17]. They showed a mixed dermal and subcutaneous infiltrate composed of mononuclear cells, neutrophils, eosinophils and even atypical myeloid cells and the occurrence of CD123-positive plasmacytoid dendritic cells as important cell population due to release of type I interferons [17]. Discussion concerning the role of plasmacytoid dendritic cells (pDC) in autoinflammatory diseases is still ongoing. pDCs are antigen presenting cells specialized for type I interferon production in response to virus or immune complexes [18]. Intriguingly, FoxP3+ regulatory T cells could also be found within their specimens. The authors interpret this as an attempt to downregulate the proinflammatory cycle in these patients [17].
In another group skin biopsies of patients with autoinflammatory disease associated with NOD2 mutations have been investigated [19]. They observed different inflammatory patterns: spongiotic dermatitis, perivascular and granulomatous dermatitis and a pigmented pruritic dermatosis. No cases of pyoderma gangraenosum or erythema nodosum have been seen [19]. In addition to erysipelas-like-eruptions in FMF, recurrent urticarial eruptions are a possible cutaneous sign of diverse systemic diseases, even autoinflammatory syndromes for example CAPS. Broeckaert and colleagues recently characterized a sensitive and specific histopathologic clue for neutrophilic urticarial dermatosis (NUD) that helps to differentiate NUD from conventional urticaria. This clue is a distinct neutrophilic epitheliotropism, neutrophilic infiltration of epidermal epithelia, hair follicles, sebaceous and sweat glands [20]. Primarily reported in 2009 by Kieffer and colleagues and later by additional working groups, NUD has been featured as a hallmark of autoinflammation [21-24]. In CAPS this feature of NUD has also been described [21,23]. A comprehensive overview on histopathologic differentials on erysipelas-like erythema is given by Kolivras and colleagues [16]. They discus infectious erysipelas, neutrophilic urticarial dermatoses, neutrophilic urticarial, urticarial vasculitis and Sweet`s syndrome. As autoinflammatory diseases may be disguised as conventional dermatoses, especially erysipelas-like rashes or common urticaria, distinct morphologic differences encompass distribution and density of the infiltrate, cell composition and cell type of the infiltrate, leukocytoclasia and changes of vessel morphology as well as absence or occurrence of dermal edema have to be investigated [16,25]. Morphologic changes are often subtle and serial sections are mandatory to diagnose NUD.
Take home messages
As a rule for clinicians, if a specific therapy does not work sufficiently, the diagnosis has to be re-evaluated. This rule applies accordingly and exceptionally for hereditary periodic fever syndromes. Intense and diligent clinical and laboratory investigations are mandatory to identify these rare patients and convey them to specific therapies. The dermatopathologist armed with distinct and copious clinical information along with histopathological examination is able to look for specific clues that lead the clinician in the direction of the diagnosis of an autoinflammatory disease.
References
- Rigante D, Frediani B, Cantarini L (2016) A Comprehensive Overview of the Hereditary Periodic Fever Syndromes. Clin Rev Allergy Immunol. [Crossref]
- Schmaltz R, Vogt T, Reichrath J (2010) Skin manifestations in tumor necrosis factor receptor-associated periodic syndrome (TRAPS). Dermatoendocrinol 2: 26-29.
- Muscari I, Iacoponi F, Cantarini L, Lucherini OM, Simonini G, et al. (2012) The diagnostic evaluation of patients with potential adult-onset autoinflammatory disorders: our experience and review of the literature. Autoimmun Rev 12: 10-13. [Crossref]
2021 Copyright OAT. All rights reserv
- Yao Q, Lacbawan F, Li J (2016) Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic. Semin Arthritis Rheum 45: 633-637. [Crossref]
- Yao Q (2013) Nucleotide-binding oligomerization domain containing 2: structure, function, and diseases. Semin Arthritis Rheum 43: 125-130. [Crossref]
- Centola M, Wood G, Frucht DM, Galon J, Aringer M, et al. (2000) The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood 95: 3223-3231. [Crossref]
- Portincasa P (2016) Colchicine, Biologic Agents and More for the Treatment of Familial Mediterranean Fever. The Old, the New, and the Rare. Curr Med Chem 23: 60-86. [Crossref]
- Aydin F, Ozcelik C, Akpolat I, Turanli AY, Akpolat T (2011) Erysipelas-like erythema with familial Mediterranean fever. J Dermatol 38: 513-515. [Crossref]
- Sohar E, Gafni J, Pras M, Heller H (1967) Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 43: 227-253. [Crossref]
- Demirkaya E, Saglam C, Turker T, KonaPaut I, Woo P, et al. (2016) Performance of Different Diagnostic Criteria for Familial Mediterranean Fever in Children with Periodic Fevers: Results from a Multicenter International Registry. J Rheumatol 43: 154-160. [Crossref]
- Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, et al. (1997) Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40: 1879-1885. [Crossref]
- Descamps V, Lejoyeux F, Marck Y, Bouscarat F, Crickx B, et al. (1997) Erysipelas-like mercury exanthem. Contact Dermatitis 36: 277-278. [Crossref]
- Durupt F, Dalle S, Ronger S, Thomas L (2006) Does erysipelas-like rash after hip replacement exist? Dermatology 212: 216-220. [Crossref]
- Ollivaud L, Ortoli JC, Saiag P, Le Frare MA, Margulis A, et al., (1993) Erysipelas-like eruption with hyperleukocytosis and fever: a new paraneoplastic syndrome in melanoma?. Ann Dermatol Venereol 120: 831-833. [Crossref]
- Goldfinger SE (1972) Colchicine for familial Mediterranean fever. N Engl J Med 287: 1302. [Crossref]
- Kolivras A, Provost P, Thompson CT (2013) Erysipelas-like erythema of familial Mediterranean fever syndrome: a case report with emphasis on histopathologic diagnostic clues. J Cutan Pathol 40: 585-90. [Crossref]
- Torrelo A, Colmenero I, Requena L, Paller AS, Ramot Y, et al. (2015) Histologic and Immunohistochemical Features of the Skin Lesions in CANDLE Syndrome. Am J Dermatopathol 37: 517-522. [Crossref]
- Santana-de Anda K, Gamez-MartÃn D, Soto-SolÃs R, Alcocer-Varela J (2013) Plasmacytoid dendritic cells: key players in viral infections and autoimmune diseases. Semin Arthritis Rheum 43: 131-136. [Crossref]
- Yao Q, Su LC, Tomecki KJ, Zhou L, Jayakar B, et al. (2013) Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol 68: 624-631. [Crossref]
- Broekaert SM, Baer-Auer A, Kerl K, Herrgott I, Schulz X, et al. (2016) Neutrophilic Epitheliotropism is a Histopathological Clue to Neutrophilic Urticarial Dermatosis. Am J Dermatopathol 38: 39-49. [Crossref]
- Herbert VG, Ahmadi-Simab K, Reich K, Baer-Auer A (2016) Neutrophilic urticarial dermatosis (NUD) indicating Cryopyrin-associated periodic syndrome associated with a novel mutation of the NLRP3 gene. J Eur Acad Dermatol Venereol 30: 852-853. [Crossref]
- Kieffer C, Cribier B, Lipsker D (2009) Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore) 88: 23-31. [Crossref]
- Kolivras A, Theunis A, Ferster A, Lipsker D, Sass U, et al. (2011) Cryopyrin-associated periodic syndrome: an autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J Cutan Pathol 38: 202-208. [Crossref]
- Lipsker D (2013) Neutrophilic urticaria with systemic inflammation identical to neutrophilic urticarial dermatosis. JAMA Dermatol 149: 1244-1245. [Crossref]
- Marzano AV, Tavecchio S, Venturini M, Sala R, Calzavara-Pinton P, et al. (2015) Urticarial vasculitis and urticarial autoinflammatory syndromes. G Ital Dermatol Venereol 150: 41-50. [Crossref]