Abstract
Introduction: The typical feature of spinocerebellar ataxia type 7 is retinal degeneration. Herein, we report an atypical spinocerebellar ataxia type 7 patient don’t have brainstem and cerebellar atrophy as well as don’t have retinal degeneration firstly.
Case presentation: The patient was 21 years old male, Han ethnic. He presented with progressive ataxia and vague speech beginning at age 15 years. In his 19 years old, he can't work normally and losing his job. His brain magnetic resonance imaging was normally, and didn’t had obvious vision loss and blindness, no memory decline.
Conclusion: Spinocerebellar ataxia type 7 is a rare genetic disorder, usually for retinal degeneration and ataxia, and vision loss usually occurs before unstable walking. But not accompanied by visual changes in patients with ataxia, we should take the gene analysis in order to avoid misdiagnosis.
Key words
spinocerebellar ataxia type 7, gene analysis, retinal degeneration
Abbreviations
SCA: Spinocerebellar Ataxia; MRI: Magnetic Resonance Imaging; ADCAII: Autosomal Dominant Cerebellar Ataxia II
Introduction
Spinocerebellar ataxia (SCA) is an autosomal dominant inherited disorder of brain function. It is characterized by increasing problems with coordination that often affect the legs, hands and speech. There are more than 20 types of SCA that have been described. Spinocerebellar ataxias are fairly rare disorders with a prevalence of 0.3 to 2 per 100, 000. SCA7 is a rare subtype of the spinocerebellar ataxias (SCAs), SCA7 accounts for 1% to 11.7% of the cases among the genetically identified conditions [1]. It is clinically characterized by ataxia, pyramidal syndrome and progressive macular dystrophy. We herein describe a family from southwest China whose proband was a 21-year- old male presenting with ataxia, and dysarthria, dysrhythmia, but he don’t have visual loss and oculomotor abnormalities, his brainstem and cerebellar don’t atrophy by magnetic resonance imaging (MRI) scan.
Case report
Index case: The patient was 21 years old male, Han ethnic, and presented with progressive ataxia and vague speech beginning at age 15 years. At the age of 15, his families and friend found that his speech was lisped and walking instability. Speech intermittently, talks on and off, unable to control speed. At the same time, he has drunk feeling when work, not his feet on the ground. At first he pay no `attention to it, but the above symptoms gradually aggravate. In his 19 years old, he can't work normally and losing his job. And have to ask for doctor, but head MRI did not see anomaly (Figure 1). No obvious vision loss and blindness, no memory decline.
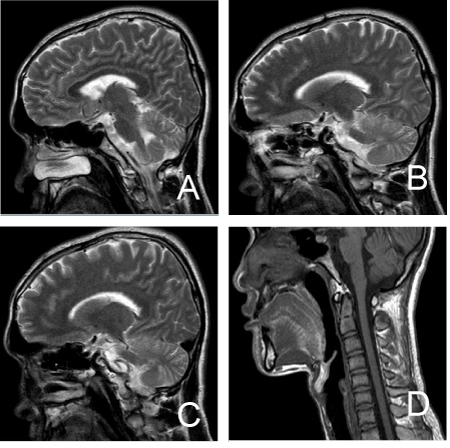
Figure 1. The brain magnetic resonance imaging of the index patient.
Neology system examination: Consciousness is clear, verbal ambiguity, recite poems sample language, intelligence is normal, vision is good, no color-blindness, gaze palsy when eye lookup, other eye movement is free, no diplopia and nystagmus. Uvula in center, gag reflex exist, Limb muscle tension normal, strength grade V, left strength was slightly weak. Tendon reflex is active, ankle clonus is positive. Double side pathological sign is positive. Left body pain is decline. Deep feeling is existed. Both finger-nose test and heel-knee-tibia test is a bit poor in two side. Upper limb alternating movement is poor, right upper limb rebound test is poor. Romberg's sign (+), linear walk can't. Double foot is cavus.
Auxiliary examination: Routine blood, blood biochemical, blood sedimentation, HIV, syphilis, tumor markers, thyroid function, hepatitis virus check all normal. Abdomen color ultrasound, Ultrasonic Cardiogram is normal. EEG is abnormal, hyperventilation induce a repeated fits 5 ~ 6 HZ θ activities. Head MRI did not see anomaly, no cerebellar or brainstem atrophy. Visual evoked potentials and Brainstem auditory evoked potentials are normal. There is normal visual field, and normal color discrimination with Ishihara color test. There was neither ophthalmoparesis nor nystagmus. Funduscopic examination showed neither macular pigmentation nor disc pallor.
The family has 15 members,and 5 members suffered form walking instability. The pedigree of the family is presented in (Figure 2). His grandmother’s father presented with walking unsteady for his old age (Specific age unknown). His grandmothers also had walking instability, and speak not clear symptoms, disease at the age of 50, 60 years old died. Her brother suffered from the same symptoms at the age of 40 years old and died in his 49. Index case’s aunt diseased with ataxia aged 12, then suicide at the age of 18.
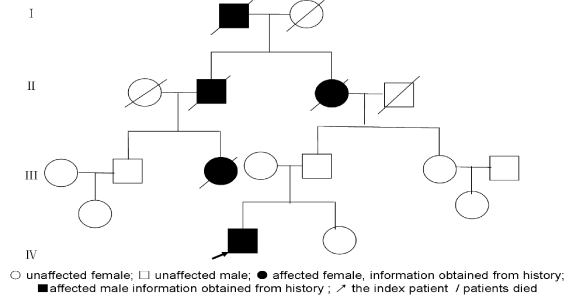
Figure 2. Family pedigrees of patients with spinocerebellar ataxia type 7
Gene analysis: The peripheral blood was collected for DNA analysis of SCA 7 mutation (CAG expansion). PCR (polymerase chain reaction) primers used for CAG expansion were 5′-TAGGAGCGGAAAGAATGTCGGAG-3′ and 5′-CAGGAAGTTTGGAAGCCTCAACC-3′. The PCR product sizes were calculated by using pBluescript sequenced with M13 primer (5′-GTTTTCCCAGTCA-CGAC3k) [2]. At the same time, DNA analysis of SCA1, SCA2, SCA 3, SCA6, SCA12 and DRPLA was taken. Genomic testing revealed a pathological expansion of CAG repeats in the SCA7 gene (repeat number=53/12). The result of our molecular analysis confirmed the clinical diagnoses of SCA7 for this family.
Discussion
Spinocerebellar ataxia type 7 (SCA 7) is a polyglutamine (polyQ) neurodegenerative disease characterized by progressive cerebellar ataxia and retinal degeneration with ophthalmoplegia, hyperreflexia, sensory loss, dysarthria, and dysphagia. In addition to performed ataxia, the most outstanding clinics characteristic is vision loss cause by retinal degeneration. Generally, vision change before ataxia. A family with Ataxia and retinal degeneration almost was SCA7 family.
Initial magnetic resonance imaging (MRI) studies on the brainstem have shown a diminished volume mainly in the cerebellum and pons, while T2 images have shown hyperintensities in transverse fibers at the pons. Neuropathological research, however, has shown more widespread brain damage including loss of myelinated fibers [3].
SCA7 had been widely reported since the advanced molecular biological technique was used to identify the related genes. SCA7 is caused by expansion of the polyQ-encoding tract of the ATXN7 gene, which encodes ataxin-7, a subunit of complexes involved in transcriptional regulation. The normal CAG repeat length is below 19, Genetic instability normal allele is 28-33, and pathogenic expanded alleles contain 37 to 460 repeats. Expansion length (greater than 37 AG repeats) is inversely correlated with age of onset and may be correlated with severity of illness [4]. Genetic testing is the gold standard of SCA7.
Our case doesn’t2021 Copyright OAT. All rights reserv, and the MRI scan didn’t find the brain stem and cerebellum atrophy. One case report from South Korea reported a patient had no retinal degeneration even in the presence of visual symptoms; the patients had a CAG repeat size of 42 and a progressive gait disturbance for 8 years [5]. But that case has a marked atrophy of the cerebellar hemispheres and 4th ventricular dilatation.
Conclusion
Herein, we report an atypical SCA7 case whose CAG repeat length is 53 diagnose by gene analysis, this patient don’t have brainstem and cerebellar atrophy, at the same time don’t have retinal degeneration for 6yr. As obtained form the index case’s father, there was no person suffered with vision loss in his family. Phenotypically, our patient had pure cerebellar syndrome without retinal degeneration, therefore was not thought to be ADCAII (autosomal dominant cerebellar ataxia II) or SCA7. Intriguingly, the index case patient don’t have neuroimaging change in cerebellar or brainstem, may be due to the short time illness. Our case again emphasizes the phenotypic variability of SCAs, and the need for genetic confirmation whenever possible.
Acknowledgement
Our paper is dedicated to the members of this family, whose personal resolve and commitment to this study are inspirational. Dr. Yang Shun performed the original genetic testing in this patient.
References
- Holmberg M, Duyckaerts C, Dürr A, Cancel G, Gourfinkel-An I, et al. (1998) Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet 7:913-8. [Crossref]
- Gu W, Wang Y, Liu X, Zhou B, Zhou Y, et al. (2000) Molecular and clinical study of spinocerebellar ataxia type 7 in Chinese kindreds. Arch Neurol 57:1513-8. [Crossref]
- Alcauter S, Barrios FA, Diaz R, Fernandez-Ruiz J. Gray and white matter alterations in spinocerebellar ataxia type 7: an in vivo DTI and VBM study. Neuroimage 55: 1-7. [Crossref]
- David G, Abbas N, Stevanin G, Dürr A, Yvert G, et al. (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17:65-70. [Crossref]
- Kim BC, Kim MK, Cho KH, Jeon BS (2002) Spinocerebellar ataxia type 7 without retinal degeneration: a case report. J Korean Med Sci 17:577-9. [Crossref]