We have previously postulated that Alzheimer’s disease (AD), mild cognitive impairment (MCI), and accelerated aging progress with similar molecular signaling and are a result of genomic vulnerability. Chronic depression–nothwithstanding comorbidties–may also enter the same pathways due to mutant alleles in the depression associated genes such as neurotransmitter-serotonin transporter proteins, e.g. 5-HTT. When depression, AD, and MCI are predisposed due to genomic vulnerability, molecular cascades for the illnesses may combine. Thus, phenotypes may reinforce each other and present an enhanced pathogenic intensity. We suggest that this model can highlight the genotypic and phenotypic interactions of these two neuropathologies and can be used to create new treatment approaches.
Alzheimer’s disease, cognitive impairment, neuropathology
We have noted that there seems to be a linked pattern in the appearance of neurobiological and clinical phenotypes for depression and AD, in earlier works [1-3]. Since neuromolecular communications are quite similar in the two pathologies, we propose that primary pathogenetic factors lie more in the genome. Although this hypothesis may create more ambiguity, it was possible for us to engender a framework for AD and depression (with or without sequelae) that uses an interaction of a particular molecular signaling series in the brain. We suggest oxidative stress as a pivotal factor.
A convenient angle to our understanding of depression is provided by Akiskal’s new psychobiologic paradigm of depressive illness [1-4], which expounds upon a “spectrum” of depressive severity (from sub-syndromal to syndromal) on which symptoms are expressed (in terms of degree or scale). As such, even sub-threshold manifestations of depression have significance and progressively aquire clinical relevance in a gradient fashion.
The paradigm holds significance in its ability to define depression as an inclusive, syndromic illness with a full spectrum of symptoms such as sub-symptomatic disease, dysthymia, sub-threshold major depression, and major depressive disorder, as well as risks such as psychosis, suicide, bipolar, and manic disorder. Influenced by Akiskal’s model, we suggest a spectrum for Alzheimer’s disease—an illness that at its inception is largely asymptomatic, sparked by genomic vulnerability, and presents few cognitive, motor, or affective impairments. One may say this is the sub-symptomatic or sub-syndromic stage on the spectrum. The sub-threshold phase could be the presence of mild cognitive stressors that progress with age.
With increasing age, cognitive and some emotional signs appear more clearly and progressively – termed aging cognitive impairment (ACI) and mild cognitive impairment (MCI), clinical stages that that have very wide ranges of intensity and may sometimes confuse physicians [5].
Lastly, the spectrum will lead to characteristic AD symptoms, which are consecutively more precarious till death. Previously we have discussed the possibility that perhaps AD is not an anomalous disorder but rather the body’s natural progression of an extended lifespan [1]. We can label that the Senility of Alzheimer Type (SAT), which is the most common clinical phenomenology of people age 88 and over [1,3,6,7]. SAT contrasts the Senility of the non-Alzheimer Type (SNAT), which is an uncommon clinical phenomenology of people age 90 and over [1,6,7]. In an effort to delineate this idea, we analyzed a fictitious sample of individuals ages 88-90 who do not die at a fixed population of 1000 (N=1000). Following general epidemiology for AD, 5% of the population bears the disease at age 65, 10-25% at 75, 20-25% at 85, and 35-50% at 100. (AD incidences in SNAT elders are discounted.) Peculiarly, at the last age group we would have an 80% probability for SAT and 20% probabilities for SNAT. Following this trend, if people lived even longer, we can extrapolate that there would be an even lower chance for SNAT. Since most individuals die between the ages of 78 and 85, data skews as a result of a changing sample size. Therefore, MCI and AD prevalence is miscalculated, which affects our perception of the nature of the two neuropathologies.
In sum, for a population of 1000 seniors age 65 without any present symptoms of AD, 800 individuals would display SAT and 200 would display SNAT by age 100. Thus, normal aging is correlated with a progression of all phenotypes—clinically, neurobiologically, and molecularly apparent—in AD. Genomic vulnerability may result in genes failing to assemble oxidative stress defense proteins, particularly AβPP, and therefore cascade into more devastating symptoms, as well as cause SAT to appear in an earlier age group (65-75 years old), which is exacerbated by comorbidities like depression. AD and MCI can be unified through the model that aging progresses alongside cognitive, psychosocial impairment [8,9]. A genomic spectrum [10,11], defined by function of RNA, DNA, enzymes, metabolism, and mitochondria, can vary in intensity and further our understanding of AD and MCI.
Pathology in depression is closely linked to AD and MCI by way of oxidative stress. 5-HTTs (serotonin transport proteins) can be defective when synthesized by mutant alleles, leading to altered reuptake [12]. Serotonin depletion leads to increased CRF/cortisol, glutamate, Ca2+, reactive oxygen species and a seven fold increased incidence of depression [13]. Free radicals enter oxidative stress pathways and individuals can develop AD or worsen a pre-existing instance of either AD or depression. If molecular signaling in depression is a form of “transverse cross-talk signaling” then signaling is a function of genomic integrity, as noted in Akiskal’s depression spectrum which may interact with pre-existing AD. Of note, neuromolecular and neurochemical signaling in patients with AD is tenuous and permanent, and depression can accelerate this process. As mentioned earlier, we propose an analogous Akiskal’s scale for AD in the order of: aging, cognitive aging disturbance, MCI, sub-threshold AD, clinical AD, and terminal AD. The natural process is in the form of “progressive longitudinal signaling,” which spans one’s lifetime, from birth to death [14]. However, genomic vulnerability, free radicals, depression, psychosocial stressors, and PTSD, among others, [12,15,16] may cause an unnatural progression of AD earlier than usual.
Phenotypically, depression may present at any age, save for infancy, and appears or disappears at various times in life [4], perhaps with the advent of second generation antidepressants like SSRI’s [13]. In indivduals ages 0-30 oxidative stress, which we previously proposed is a major factor for AD and depression, is controlled by neuro-antioxidant defense systems, such as enzymes, vitamins, and chaperone proteins [14].
However, in the middle ages of 30-60, metabolic processes, cellular and molecular signaling in the mitochondria [17], and DNA/RNA repair without transcription bias play a role in healthy metabolism and preventing oxidative stress. As these pathways become more prone to error, reactive oxygen and nitrogen species increase in concentration, which further damage mitochondrial DNA, RNA, neuronal and glial proteins, lipids and carbohydrates, all of which are exacerbated in the presence of genomic vulnerability [18]. Neural antioxidant defense agents and buffers increase resistance to damage from oxidative stress to valuable cellular machinery. Particularly, AβPP, PS1, PS2, tau, and ApoEε4 increase in concentration in the presence of oxidative stress [19,20]. Malfunction of these proteins, which depression may help propagate, leads to the formation of senile plaques and neurofibrillary tangles, a tell-tale sign of AD [21]. However, it is not until the third age group of 60-90 years old that the AD phenotypes appear [20].
In the 60-90 year old age group, clinically relevant symptoms begin to appear. Specific to one’s proclivity for antioxidants in the brain among redox metals, Aβ peptide, hyperphosphorylated tau, and ApoEε4 may be deposited in or around neurons leading to senile plaques [22,23]. There is evidence that the hippocampus [24] and frontal cortex are particularly atrophied by these neurochemical changes brought upon by oxidative stress. Likewise, protease and phosphorylation errors primarily lead to malfunctioning AβPP [19], PS1, PS2, tau, and ApoE [25,26]. In depression, genomic vulnerability [27] enhances this neuromolecular misprocessing in the elderly by interacting with AD signaling pathways that speed up increasing glutamate and Ca2+ concentrations, neurodegeneration of the hippocampus, and oxidative stress [15,16,27-31] ( Figure 1).
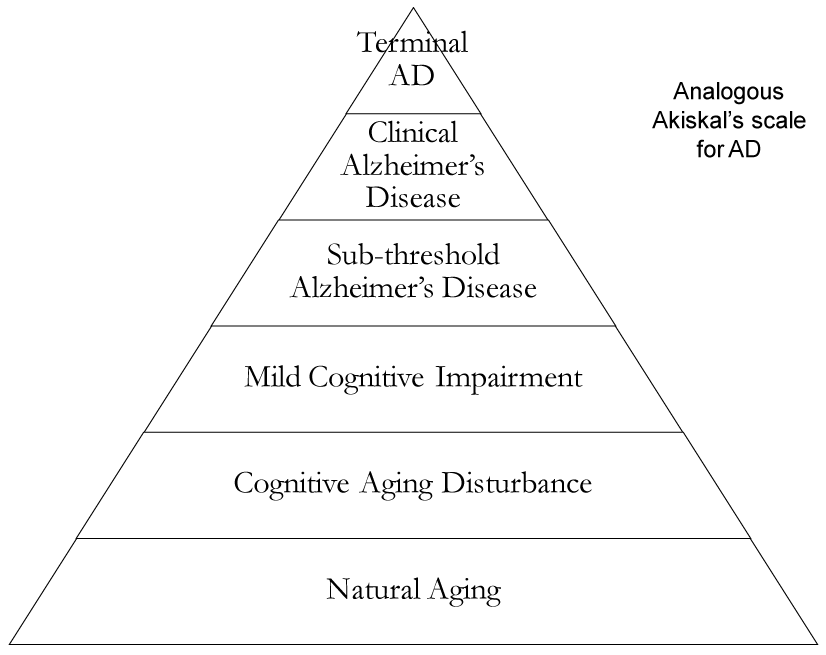
Figure 1. Diagram illustrating the transition of natural aging to terminal AD though sequential transformation based on Akiskal’s scale
Our suggested model of AD, and its relationship with aging, is more in tune with clinical observations in geriatric patients who present with mild memory impairment but a full range of daily living activites. Without a definitive ability to check for AD till post-mortem [32], this model aims to provide a scale for accurate diagnoses early on.
If a geriatric patient has had a history of depression [33] and memory deficits, then it is a stronger indicator of latent AD. Especially in the case of chronic depression, genomic vulnerability for AD will increase, so preventative measures should be taken before debilitating symptoms appear. If a patient is a carrier of AD, then the physician should scan for attenuated symptoms along the “depressive cross-talk scale,” and may have to employ measures for activated AD or resultant dementia. The treatment team can then find contraindicators for accelerated aging, MCI, and classic chronic depression.
Treatment of other depressive episodes at any stage in one’s life, such as MDD, should also be considered as indicators of a quicker onset of AD. Traumatic brain injury, PTSD, accelerated aging [34], and psychosocial decline can be addressed with cognitive behavioral therapy, lifestyle changes, and medications. A psychiatrist may recommend antidepressant SSRI’s, among others, while a nutrionist may recommend a change to a Mediterranean diet.
Our suggested model consistently addresses accelerated aging, MCI, and AD by tying depression pathways and AD pathology together. Diagnostic imaging studies also support this model as soon as there is significance in the reslts. We firmly hold that future research focused on this model can provide a better understanding of the nature of these neuropathologies and will shape prevention in the future.
- Rodrigues R, Bonda DJ, Perry G, Castellani RJ, Casadesus G, et al. (2010) Neurodegeneration: An Inevitable Consequence of Aging? Implications for Therapy. in Michael S. Ritsner (Editor) Brain Protection in Schizophrenia, Mood and Cognitive Disorders Oxidative Stress. Springer: 305-324.
- Rodrigues R, Smith MA, Wang X, Perry G, Lee HG (2012) Molecular neuropathogenesis of Alzheimer’s disease: an interaction model stressing the central role of oxidative stress. Future Neurol 7: 287–305. [Crossref]
- Rodrigues R, Petersen RB, Perry G (2014) Parallels between major depressive disorder and Alzheimer’s disease: role of oxidative stress and genetic vulnerability. Cell Mol Neurobiol 34: 925-949. [Crossref]
- Akiskal HS, Judd LL (2005) The depressive spectrum: reconceptualizing the relationship between dysthymic, subthresold and Major depressions 47-70.in Licinio J., Ma-Li Wong Biology of Depression. From Novel Insights to Therapeutics Strategies. WILWY – VCH Verlag: 47-70.
- Daffner KR, Scinto LFM (2000) Early diagnosis of Alzheimer’s disease: an introduction. In: Daffner KR, Scinto LFM (eds) Early Diagnosis of Alzheimer’s Disease. Humana Press, Inc., Totowa, NJ: 1–28: 925–949.
- Azari NP, Pettigrew KD, Schapiro MB, Haxby JV, Grady CL, et al. (1993) Early detection of Alzheimer’s disease: a statistical approach using positron emission tomographic data. J Cereb Blood Flow Metab 13: 438-447.[Crossref]
- Bachman DL, Wolf PA, Linn RT, Knoefel JE, Cobb JL, et al. (1993) Incidence of dementia and probable Alzheimer’s disease in a general population: the Framingham Study. Neurology 43: 515-519. [Crossref]
- Vijg J (2007) Aging of the Genome: the Dual Role of the DNA in Life and Death. Oxford University Press, Oxford.
- Wilson DM 3rd, Bohr VA, McKinnon PJ (2008) DNA damage, DNA repair, ageing and age-related disease. Mech Ageing Dev 129: 349-352. [Crossref]
- Moreira PI, Nunomura A, Honda K, et al. (2007) The key role of oxidative stress in Alzheimer’s disease. In: Qureshi GA, Parvez SH (eds) Oxidative Stress and Neurodegenerative Disorders. Elsevier B.V., Amsterdam: 267-281.
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, et al. (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60: 759-767. [Crossref]
- Canli T, Omura K, Haas BW, Fallgatter A, Constable RT, et al. (2005) Beyond affect: a role for genetic variation of the serotonin transporter in neural activation during a cognitive attention task. Proc Natl Acad Sci U S A 102: 12224-12229. [Crossref]
- Del Rio J, Frencilla D (2005)2021 Copyright OAT. All rights reservt WJ, Reith MEA (eds) Dopamine and Glutamate in Psychiatric Disorders. Humana Press, Totowa, NJ: 215-234.
- Heininger K (2000) A unifying hypothesis of Alzheimer’s disease. III. Risk factors. Hum Psychopharmacol 15: 1-70. [Crossref]
- McEwen BS (1999) Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105-122. [Crossref]
- Kendler KS, Karkowski LM, Prescott CA (1999) Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156: 837-841. [Crossref]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, et al. (1999) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19: 1959-1964. [Crossref]
- Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36: 747-751. [Crossref]
- Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, et al. (2003) Amyloidbeta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 43: 1-16. [Crossref]
- Cappai R, Needham BE, Ciccotosto GD (2007) The function of the amyloid precursor protein family. In: Collin JB, David HS (eds) Abeta Peptide and Alzheimer’s Disease: Celebrating a Century of Research. Springer, London; 37-51.
- Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, et al. (1993) Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 342: 697-699. [Crossref]
- Baum L, Chen L, Ng HK, Pang CP (2000) Apolipoprotein E isoforms in Alzheimer’s disease pathology and etiology. Microsc Res Tech 50: 278-281. [Crossref]
- Weisgraber KH, Pitas RE, Mahley RW (1994) Lipoproteins, neurobiology, and Alzheimer’sdisease: structure and function of apolipoprotein. E Curr Opin Struct Biol 4: 507-515.
- Smith MA, Rodrigues R (2009) The twin frontiers of depression and Alzheimer’s disease. Front. Neurosci 3, 236–237.
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, et al. (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: 15-20. [Crossref]
- Richey PL, Siedlak SL, Smith MA, Perry G (1995) Apolipoprotein E interaction with the neurofibrillary tangles and senile plaques in Alzheimer disease: implications for disease pathogenesis. Biochem Biophys Res Commun 208: 657-663. [Crossref]
- Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, et al. (2006) Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 65: 631-641. [Crossref]
- Wong ML, Licinio J (2001) Research and treatment approaches to depression. Nat Rev Neurosci 2: 343-351. [Crossref]
- Sapolsky RM (2000) The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry 48: 755-765. [Crossref]
- Smith MA, Makino S, Kvetnansky R, Post RM (1995) Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15: 1768-1777. [Crossref]
- Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, et al. (2006) Increased hippocampal plaques andtangles in patients with Alzheimer disease witha lifetime history of major depression. Arch Gen Psychiatry 63: 161–167. [Crossref]
- Sharma HS, Westman J, Cervós-Navarro J, Dey PK, et al. (1997) Opioid receptor antagonists attenuate heat stress-induced reduction in cerebral blood flow, increased blood-brain barrier permeability, vasogenic edema and cell changes in the rat. Ann N Y Acad Sci 813: 559-571.
- Freeman T, Roca V, Guggenheim F, Kimbrell T, Griffin WS (2005) Neuropsychiatric associations of apolipoprotein E alleles in subjects with combat-related posttraumatic stress disorder. J Neuropsychiatry Clin Neurosci 17: 541-543. [Crossref]
- Porter NM, Landfield PW (1998) Stress hormones and brain aging: adding injury to insult? Nat Neurosci 1: 3-4. [Crossref]
Editorial Information
Editor-in-Chief
Masayoshi Yamaguchi
Emory University School of Medicine
Article Type
Research Article
Publication history
Received: December 16, 2014
Accepted: March 12, 2015
Published: March 17, 2015
Copyright
©2015 Rodrigues R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
Rodrigues R, Ahmad F, Perry G, Petersen R (2015) Synergy between depression and Alzheimer’s disease: A spectrum model of genomic vulnerability with therapeutic implications. Integr Mol Med 2: DOI: 10.15761/IMM.1000128