Key words
imidazoline ring, bioversatile scaffold, receptor systems, morphine dependence, antidepressant agents
The first drug bearing the 2-substituted imidazoline ring as a structural motif was the α-adrenoreceptor (α-AR) agonist nafazoline [1], described in 1939 and still used as vasoconstrictor and nasal decongestant. Twenty-five years later Boehringer Ingelheim patented clonidine [2], a drug able to cause high sedation, reduction of blood pressure and cardiac rate, and inhibition of salivary secretion. It also showed analgesic properties useful in the treatment of neuropathic pain. Its central sympathetic inhibition was ascribed to the activation of the α2-ARs in the medulla oblongata. However, in 1984 Bousquet hypothesized a complementary mechanism involving the I1-imidazoline binding sites (I1-IBS) [3]. Though IBS, also including I2 and I3 subtypes, are distinct from α2-ARs, they similarly recognize ligands bearing the imidazoline ring. The discovery of the vasoconstrictor cirazoline (1) (Figure 1), emerged as an α1-AR agonist/α2A-AR antagonist/α2C-AR agonist and efficacious I2-IBS ligand [4], supported the versatility of the imidazoline nucleus as a structural motif of molecules with different biological profiles [5].
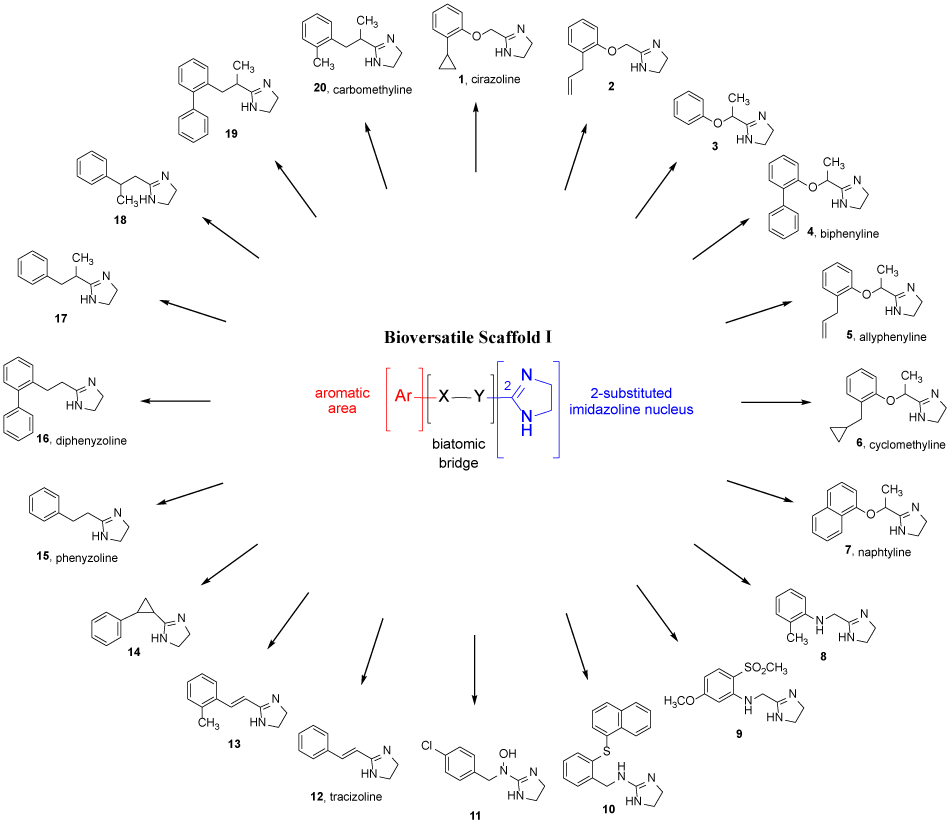
Figure 1. Chemical structures of compounds 1-20 sharing the common scaffold I.
In the structure of cirazoline, as well as of many other active ligands directed to different biological systems, it is possible to recognize the bioversatile scaffold I reported in Figure 1, consisting of the 2-substituted imidazoline nucleus linked to an aromatic area (Ar) by a biatomic bridge (X-Y). Interestingly, the three portions forming such a scaffold display different functions. Pigini e coll. noticed that minor chemical modifications in the biatomic bridge determined the preferential recognition of one or more specific biological targets [6], whereas those in the aromatic moiety (in particular in the ortho position of the phenyl ring) were generally responsible for the functional behaviour of the ligand [7].
In particular, the -OCH2- chain is compatible with ligands directed to I2-IBS and α2-ARs. Indeed, several cirazoline analogues have been described as multitarget agents, producing efficacious α2C-AR agonism/α2A-AR antagonism/I2-IBS interaction. In vivo experiments performed with compound 2 demonstrated that such a combination is useful for reducing opioid withdrawal syndrome and associated depression [8].
The introduction of a methyl group in the oxymethylene chain, leading to the -OCH(CH3)- bridge, proved to be particularly suitable for the α2-ARs. Indeed, such a modification drastically reduced the I2-IBS affinity. In particular, compound 3 behaved as a potent α2-AR antagonist and showed high selectivity over IBS [6]. Interestingly, it has been observed that the insertion of an ortho phenyl substituent in the aromatic ring of 3 modulates its fuctional profile. Indeed, biphenyline (4) is characterized by an α2-AR agonist profile. The eutomer S-(-)-4, tested in two algesiometric tests (rat hot-plate and tail-flick), displayed a potent and long lasting antinociceptive effect, clearly mediated by α2-ARs, as it was completely blocked by the selective α2-antagonist RX 821002 [9].
The results obtained with biphenyline stimulated the investigation of the effect produced by the insertion of different substituents in the ortho position of the aromatic ring of the antagonist 3. In particular, substituents such as cyclopentyl or cyclohexyl groups, endowed with steric bulk comparable to that of the phenyl ring, led to compounds behaving similarly to 4 (agonists at both α2A- and α2C-AR subtypes). On the contrary, substituents such as allyl or cyclopropyl groups, with lower steric bulk (MR <20), induced significant modulation of the antagonist profile of 3 only at the α2C-subtype. Indeed, they maintained the α2A-antagonist behavior, but were potent α2C-agonists [7]. This last in vitro biological profile proved to be responsible for the intriguing pharmacological behavior of the allyl derivative 5 (allyphenyline). Indeed, in in vivo studies in mice, allyphenyline enhanced morphine analgesia (due to its a2C-agonism), was devoid of sedative effect (due to its a2A-antagonism) [4], contrasted and prevented morphine tolerance and dependence at the very low dose of 0.05 mg/kg [10]. Moreover, allyphenyline significantly reduced hyperanxiety-like behavior verified 6 days after alcohol intoxication and hyperlocomotor activity associated with alcohol withdrawal [11].
The evaluation of a large number of α2-AR ligands, including the potent α2A-AR antagonist/α2C-AR agonist cyclomethyline (6), suggested that their α2A-AR agonist or antagonist profile might be ascribed to different conformations adopted by the molecules. In particular, from a structural overlay investigation it emerged that a preferred extended conformation appeared to be associated with dual α2C-AR agonism/α2A-AR antagonism, whereas a folded conformation was associated with α2C-/α2A-AR agonism. This latter conformation is probably induced by the electron-rich ortho phenyl substituent, able to provide a stable π-cation like charge transfer complex with the imidazoline moiety [12].
Further studies performed with allyphenyline (5) and cyclomethyline (6) demonstrated that their efficacy against morphine dependence [10,13] proved to be even improved by their ability to reduce the depressed mood, partially due to the additional serotonin 5-HT1A receptor activation. For this reason, at the same dose of 0.05 mg/kg 5 and 6 exerted a potent antidepressant-like effect in the mouse forced swimming test. Experiments carried out in the presence of the α2-AR antagonist yohimbine and the serotonin 5-HT1A receptor antagonist WAY100135 demonstrated that both α2C-AR and 5-HT1A receptor activation was involved in their antidepressant-like activity [13]. Moreover, the therapeutic potential of 5 and 6 was strongly enhanced by their favorable in vitro ADME profiles and limited activity on the hERG channel [13].
Recently, the analogue (S)-(+)-7 [(S)-(+)-naphtyline] proved to exert an even more efficacious antidepressant-like effect in the mouse forced swimming test, being active at the very low dose of 0.01 mg/kg. This effect was also clearly mediated by the 5-HT1A receptor, as it was significantly reduced by the pretreatment with the antagonist WAY100635 [14].
The isosteric substitution of the oxygen in the bridge with an NH function, together with the insertion of suitable substituents in the ortho position of the aromatic moiety also led to biologically active compounds. Indeed, the -NH-CH2- chain proved to be suitable for ligands interacting with I1-, I2-IBS and α2-ARs. Among all the ligands, 8 proved to be effective in reducing expression and acquisition of morphine dependence in mice at the dose of 5 mg/kg and, therefore, might be considered a promising tool in managing opioid addiction [15].
The -NH-CH2- bridge also demonstrated to be compatible with α1-AR agonist activity, with compound 9 showing high selectivity for α1a-AR over α1b- e α1d-AR (250 and 7000 fold, respectively) [16].
The reversed bridge -CH2NH- is also present in several ligands, among which compound 10 showed significant in vitro antiplasmodial activity against both chloroquine-resistant P. falciparum W2 and atovaquone-resistant P. falciparum FCR3 strains [17].
The insertion of an hydroxy group at the nitrogen led to the -CH2N(OH)- bridge, present in a series of N-(Imidazolidin-2-ylidene)-1-arylmethanamine oxides that bind to α-ARs and IBS. Particularly interesting was the derivative 11, which showed high affinity for I2-IBS and good selectivity over I1-IBS and α-ARs. Due to low lipophilicity, such a compound is not expected to cross the blood brain barrier and, therefore, might find application for the treatment of vascular hyperplasia [18].
Structure-activity relationship (SAR) studies also demonstrated that a wholly carbon bridge is responsible for effective selectivity for IBS over α-ARs. For example tracizoline (12), a ligand bearing a -CH=CH- bridge, is endowed with high affinity for both I1- and I2-IBS and good selectivity over both α1- and α2-ARs [6]. Such a biological profile is responsible for the ability of the tracizoline analogue 13 to contrast and prevent morphine dependence in mice at the dose of 5 mg/kg [15].
A selectivity between I1- and I2-IBS subtypes can be obtained by replacing the vinyl bridge with a cyclopropyl ring. In particular, the trans isomer of 14 showed higher affinity than the cis one for both I1- and I2-IBS, indicating that the distance between the phenyl ring and the imidazoline ring is a critical requirement for the affinity. The most important result was obtained by resolving the racemate of the trans isomer into the enantiomers (1R,2R)-(-)-14 and (1S,2S)-(+)-14. At I1-IBS the dextrorotatory enantiomer was more potent than the corresponding optical antipode. At I2-IBS, enantioselective binding was also observed, but the eutomer was the levororotatory enantiomer. This was clearly a reversal of enantioselectivity with the (1S,2S)-(+)-14 being 10 fold selective for I1-IBS while the enantiomer (1R,2R)-(-)-14 about 60-fold selective for I2-IBS [19].
Further SAR studies demonstrated that the -CH2CH2- bridge was suitable for the I2-IBS interaction. It is present in the ligand phenyzoline (15), showing very high selectivity for I2-IBS over both α2-ARs and I1-IBS (I2/α2 = 794; I2/I1 = 1479) [6]. Analogously to what observed with α2-ARs derivatives, the ortho-phenyl substitution also affected the functional profile of I2-IBS ligands. Indeed, 15 and its ortho-phenyl derivative 16 (diphenyzoline) highlighted an interesting “positive” or “negative” morphine analgesia modulatory effect, respectively. In fact, 15 (s.c. 10 mg/kg) enhanced morphine analgesia, while 16 (s.c. 10 mg/kg) decreased it [20]. A subsequent study of two series of 2-aryl-ethylen-imidazolines, inspired by 15 and 16, confirmed the interesting “positive” or “negative” morphine analgesia modulation displayed by their corresponding leads and demonstrated that these effects might be correlated with morphine tolerance and dependence, respectively [21].
The α- or β-methylation of 15 appeared extremely critical regarding the affinity and selectivity for I1- or I2-IBS interaction. Indeed, the α-methylation (compound 17) favoured the I1-IBS selectivity (I1/I2 = 186) and caused high stereospecificity of interaction (eudismic ratio (S)-(-)-17/(R)-(+)-17 = 5888). Instead, even if at a lesser extent, the β-methylation (compound 18) favored I2-IBS selectivity (I2/I1 = 50) [6].
The insertion of a phenyl ring in the ortho position of 17, leading to 19, induced an interesting modulation of its biological profile from antagonism to agonism. Indeed, 19, behaving as efficacious I1-IBS agonist, produced significant hypotensive and bradicardic effects, prevented by the pre-treatment with 17. Moreover, chirality appeared crucial in the activation of the I1-IBS as the biological effects shown by 19 were produced only by the S-(+) enantiomer, while the distomer R-(-) was devoid of hemodynamic action [22]. Ten years later, the preparation of other ligands carrying selected substituents in the ortho position of the phenyl ring led to carbomethyline (20). This derivative and its (S)-(+) eutomer, showed a hypotensive effect higher than that evoked by 19 and selectively modulated by I1-IBS [23]. At the same time, this study confirmed that the introduction of ortho substituents with moderate steric hindrance and positive lipophilic contribution was able to induce antagonism to agonism modulation.
Altogether, the results described in this review strongly support the bioversatility of the imidazoline scaffold I, whose decorations can lead to selective or multitarget ligands. In the latter case, as observed for 5 and 6, the biological profile can be potentially useful for the management of comorbidities. Therefore, the 2-substituted imidazoline ring linked to an aromatic moiety by a biatomic bridge might be considered a privileged structure and deserves the ironical definition of “biological picklock”.
Acknowledgement
Dedicated to Prof. Maria Pigini, who passed away on February 8th, 2016
References
- Sonn A (1939) U.S. 2.161.938. Chem Abstr 39: 7316.
- Zeile K, Hauptmann KH, Stahle H (1965) U.S. 3.202.660. Chem Abstr 64: 11518.
- Bousquet P, Feldman J, Schwartz J (1984) Central cardiovascular effects of alpha-adrenergic drugs: differences between catecholamines and imidazolines. J Pharmacol Exp Ther 230: 232-236.
- Cardinaletti C, Mattioli L, Ghelfi F, Del Bello F, Giannella M, et al. (2009) Might adrenergic a2C-agonists/a2A-antagonists become novel therapeutic tools for pain treatment with Morphine? J Med Chem 52: 7319-7322.
- Krasavin M (2015) Biologically active compounds based on the privileged 2-imidazoline scaffold: the word beyond adrenergic/imidazoline receptor modulators. Eur J Med Chem 97: 525-537.
- Gentili F, Bousquet P, Brasili L, Dontenwill M, Feldman J, et al. (2003) Imidazoline Binding Sites (IBS) profile modulation: key role of the bridge in determining I1-IBS or I2-IBS selectivity within a series of 2-phenoxymethylimidazoline analogues. J Med Chem 46: 2169-2176.
- Gentili F, Cardinaletti C, Vesprini C, Carrieri A, Ghelfi F, et al. (2008) Alpha2-adrenoreceptors profile modulation. 4. From antagonist to agonist behavior. J Med Chem 51: 4289-4299. [Crossref]
- Del Bello F, Diamanti E, Giannella M, Mammoli V, Mattioli L, et al. (2013) Exploring multitarget interactions to reduce opiate withdrawal syndrome and psychiatric comorbidity. ACS Med Chem Lett 4: 875-879.
- Gentili F, Bousquet P, Brasili L, Caretto M, Carrieri A, et al. (2002) Alpha2-adrenoreceptors profile modulation and high antinociceptive activity of (S)-(-)-2-[1-(biphenyl-2-yloxy)ethyl]-4,5-dihydro-1H-imidazole. J Med Chem 45: 32-40. [Crossref]
- Del Bello F, Mattioli L, Ghelfi F, Giannella M, Piergentili A, et al. (2010) Fruitful adrenergic α2C-agonism/α2A-antagonism combination to prevent and contrast morphine tolerance and dependence. J Med Chem 53: 7825-7835.
- Ubaldi M, Del Bello F, Domi E, Pigini M, Nasuti C (2015) Investigation of allyphenyline efficacy in the treatment of alcohol withdrawal symptoms. Eur J Pharmacol 760: 122-128. [Crossref]
- Diamanti E, Del Bello F, Carbonara G, Carrieri A, Fracchiolla G, et al. (2012) Might the observed α2A-adrenoreceptor agonism or antagonism of allyphenyline analogues be ascribed to different molecular conformations? Bioorg Med Chem 20: 2082-2090. [Crossref]
- Del Bello F, Diamanti E, Giannella M, Mammoli V, Marchioro C, et al. (2012) Low doses of allyphenyline and cyclomethyline, effective against morphine dependence, elicit antidepressant-like effect. ACS Med Chem Lett 3: 535-539.
- Del Bello F, Cilia A, Carrieri A, Fasano DC, Ghelardini C, et al. (2016) The Versatile 2-Substituted Imidazoline Nucleus as a Structural Motif of Ligands Directed to the Serotonin 5-HT1A Receptor. Chem Med Chem 11: 2287-2298. [Crossref]
- Giusepponi ME, Cifani C, Micioni Di Bonaventura MV, Mattioli L, Hudson A, et al. (2016) Combined interactions with I1-, I2-imidazoline binding sites and α2-adrenoreceptors to manage opioid addiction. ACS Med Chem Lett 7: 956-961.
- Hodson SJ, Bishop MJ, Speake JD, Navas F, Garrison DT, et al. (2002) 2-(Anilinomethyl)imidazolines as α1 adrenergic receptor agonists: the discovery of α1a subtype selective 2’-alkylsulfonyl-substituted analogues. J Med Chem 45: 2229-2239.
- Carrasco MP, Gut J, Rodrigues T, Ribeiro MH, Lopes F, et al. (2013) Exploring the Molecular Basis of Qo bc1 Complex Inhibitors Activity to Find Novel Antimalarials Hits. Mol Inform 32: 659-670. [Crossref]
- Saczewski J, Hudson A, Laird S, Rybczyńska A, Boblewski K, et al. (2012) N-(Imidazolidin-2-ylidene)-1-arylmethanamine oxides: synthesis, structure and pharmacological evaluation. Arch Pharm (Weinheim) 345: 33-42. [Crossref]
- Quaglia W, Bousquet P, Pigini M, Carotti A, Carrieri A, et al. (1999) L. 2-(2-Phenylcyclopropyl)imidazolines: Reversed Enantioselective Interaction at I1 and I2 Imidazoline Receptors. J Med Chem 42: 2737-2740.
- Gentili F, Cardinaletti C, Carrieri A, Ghelfi F, Mattioli L, et al. (2006) Involvement of I2-imidazoline binding sites in positive and negative morphine analgesia modulatory effects. Eur J Pharmacol 553: 73-81. [Crossref]
- Gentili F, Cardinaletti C, Vesprini C, Ghelfi F, Farande A, et al. (2008) Novel ligands rationally designed for characterizing I2-imidazoline binding sites nature and functions. J Med Chem 51: 5130-5134. [Crossref]
- Gentili F, Bousquet P, Carrieri A, Feldman J, Ghelfi F, et al. (2005) Rational Design of the New Antihypertensive I1-Receptor Ligand 2-(2-Biphenyl-2-yl-1-methyl-ethyl)-4,5-dihydro-1H-imidazole. Lett Drug Des Disc 2: 571-578.
- Del Bello F, Bargelli V, Cifani C, Gratteri P, Bazzicalupi C, et al. (2015) Antagonism/Agonism modulation to build novel antihypertensives selectively triggering I1-imidazoline receptor activation. ACS Med Chem Lett 6: 496-501. [Crossref].