Abstract
Breast cancer is a complex disease with different phenotypes associated with genetic and non-genetic risk factors. An aberrant expression of the BRCA1 tumor
suppressor as well as dysfunction of BRCA1 protein caused by germline mutations are implicated in breast cancer aethiology. BRCA1 plays a crucial role in genome
and epigenome stability. Its expression is auto regulated and modulated by various cellular signals including metabolic status, hypoxia, DNA damage, estrogen
stimulation. The review describes breast cancer risk factors, the BRCA1 gene expression and functions as well as covers the role of long-range genomic interactions,
which emerge as regulators of gene expression and moderators of genomic communication. The potential long-range interactions of the BRCA1 promoter (driven by
polymorphic variant rs11655505 C/T, as an example) and their possible impact on the BRCA1 gene regulation and breast cancer risk are also discussed.
Key words
BRCA1, breast cancer, long-range genomic interactions, transcriptional regulation, stem cells
Introduction
Female breast cancer is one of the most common cancers worldwide
with app.1.68 million cases diagnosed annually [1]. It is a complex
disease characterized by molecular and phenotypic heterogeneity
observed both within populations and intra-individually, within
single tumor cells in a spatiotemporal manner, as pointed out by
[2]. This intra-individual molecular diversity could be reflective of
the mutational history of tumor cells. It is hypothesized that breast
cancer can result from clonal expansion of adult stem cells and/or stem
progenitor cells, which became cancer stem cells by acquiring tumor
initiating capacity [3].
Genetic and non-genetic breast cancer risk factors
A minority of breast cancers (5-10% of all cases) demonstrate
familial clustering and have an important genetic component [4].
Familial relative risk (FRR) increases progressively along with the
number of affected relatives [5]. Family history is influenced by a
number of complex genetic mechanisms, including prenatal effects,
mitochondrial variants, sex-liked genes and parental of origin effects
exerted by imprinted genes. These may cause its asymmetry and skew
the risk of breast cancer towards maternal lineage [6,7]. To date, genetic
factors underlying the disease are not fully elucidated.
Rare, high-risk mutations in BRCA1/BRCA2 genes account for less
than 20% of FRR. The penetrance of these mutations is incomplete,
which suggests there may be modifiers of breast cancer risk among
carriers of BRCA1/BRCA2 mutations [4]. To date, a total of 26 and
16 single nucleotide polymorphisms (SNPs), bearing a small risk (in
range 1.05-1.26) have been discovered for carriers of these mutations
by genome-wide association study (GWAS). Many of them are
associated with estrogen receptor (ER) status of the tumor subtype,
reviewed by [8]. The risk of breast cancer among BRCA1 mutations
carriers is supposed to be influenced by polymorphic variants on the
wild-type BRCA1 allele. This could possibly occur through altering the
efficiency of BRCA1 transcription [9]. However, to date the mechanism
underlying effect of the BRCA1 promoter variant rs11655505 C>T
remains unknown.
Besides high risk BRCA1/BRCA2 mutations, moderate risk
mutations such as those found in DNA repair genes (CHEK2, ATM,
PALB2) also demonstrate familial clustering. They explain 2-5% of the
FRR [10].
Common low risk SNPs identified by GWAS account for even
smaller share of FRR. Similarly to variants identified among BRCA1/
BRCA2 mutations carriers, common SNPs display differences in genetic
susceptibility to ER positive and ER negative tumor subtypes. This
suggests that common mechanisms may underlie these phenotypes [8].
In summary, polymorphic variants SNPs identified to date (more
than 70), their multiplicative effects (modeled as a polygenic score
[PRR]), taken together with mutations in BRCA1, BRCA2, PALB2,
ATM, CHEK2 genes account for one-third of FRR.
Based on the data, factors identified to date, do not fully explain
genetic susceptibility that is indicated by family history and heritability
evidence from studies on monozygotic twins [6,10-13].
Numerous genetic variants with even lower effects on risk, omitted by GWAS, might account for missing heritability. A novel approaches
has been recently proposed, involving fine SNPs mapping for subthreshold
loci or of reanalyzing and validating GWAS results by using
epigenomic signatures [14,15].
Gaining insight into biological function of SNPs is particularly
challenging because over 95% of the identified genetic variants fall
into non-coding genomic regions and three-quarters of them associate
with DNAse I-hypersensitive sites. This suggests that they lie within
regulatory elements, known to establish long-range contacts and affects
target genes located distally even several megabases away. Recently,
a number of regulatory SNPs were assigned to their target in breast
cancers, when SNPs long-range contacts were taken into account. This
was done through linking GWAS and Hi-C analyses (whole genome
conformation analysis based on proximity ligation followed by highthroughput
sequencing), reviewed by [16].
Especially noteworthy is the evidence, that the majority of breast
cancer cases (90-95%) occur sporadically [17]. This may be linked to
many factors including gender, age, reproductive and hormonal history,
environmental exposure and/or life style (alcohol intake, tobacco
smoking, diet habits (specifically high fat diet and toxine exposure)
and other stress factors [8,18,19]. Interestingly, several disorders such
as obesity and associated metabolic syndrome (including diabetes) are
reported to be related to breast cancer risk [20,21]. Similarly to the risk
of breast cancer, the risk of obesity and metabolic disorders may be
sexually dimorphic- presumably modulated by gonadal hormones and
by sex chromosome status (XX, XY) [22]. The broad spectrum of breast
cancer risk factors and related disorders has been comprehensively
discussed by [8,18].
BRCA1ness in sporadic breast cancers
BRCA1 tumor suppressor is implicated in aetiology of both familial
and sporadic breast cancer. Almost 33% of non-familial, invasive
sporadic breast cancer either lack or have a reduced expression of
BRCA1 (due to somatic alternation or epigenetic silencing) and share
the familial-BRCA1 mutated tumor’s phenotype, as reviewed recently
by [23]. The loss of BRCA1 or its dysfunction is presumably the critical
step for the formation of the basal-like subtype of breast cancer (BBC), a
high-grade, aggressive tumor with lymphocytic infiltrates. The minority
of cases (10-30%) show hypermethylation of the BRCA1 promoter [24].
Their transcriptomic signature is characterized by expression of genes
mostly active in breast myoepithelial layer (basal-layer) [23], whereas
their epigenetic characteristic is similar to those observed in embryonal
stem cells (esc), including overexpression of pioneer transcription
factors (Nanog, SOX2 and c-Myc) and under-expression of Polycombregulated
genes, as reported by [25].
A significant portion of sporadic breast cancer are estrogen receptor
negative (ER-), similarly to familial BRCA1 mutated tumors. They tend
to lack progesterone receptor (PR-), ERB-2 oncogen (HER2) and display
triple-negative TN phenotype. Molecular characteristic of these tumors
also includes genomic and chromosomal instability as stated by [26,27].
Chromosome X gains were observed in neoplastic transformation of
male epithelial cells [28], whereas epigenetic instability/loss of inactive
X chromosome frequently occurs in female basal breast cancer cases
[29]. Similarly to other solid tumors, basal like tumors demonstrate
heterogeneity of their tumoral microenviroment, including intratumoral
level of oxygen, nutrient and pH [30,31].
Hypoxia stress has been found to induce down-regulation of
BRCA1 expression and this partially explains repression of the BRCA1
gene observed in sporadic breast cancers [32]. Due to the cellular
role of BRCA1, its deficiency may contribute to genomic instability
and predispose cells to high risk of malignant transformations.
Furthermore, the expression of BRCA1 has been reported to be
required for differentiation of breast stem cells, specifically luminal
progenitor cell (ER-negative) to mature luminal cells (ER-positive). A
loss of BRCA1 may result in the accumulation of genetically unstable
breast stem cells, which presumably underlie the aetiology of basallike
breast cancers as stated by [33]. Notably, exposure to hypoxia
has recently been demonstrated to induce breast cancer stem cells
phenotype [34].
Transcriptional regulation of BRCA1 expression
As evidence emerged, that BRCA1 expression is downregulated
in sporadic breast tumors many aspects of its regulation has been
extensively studied.
The BRCA1 gene is transcribed from the bidirectional promoter
for BRCA1 and for lncRNA NBR2 (Neighbor of BRCA1 Number 2)
separated by short (approximately 218 bp) intergenic region [35,36].
This transcriptional unit arose in result of segmental duplication
during primate evolution [37]. In mice the BRCA1 gene shares
bidirectional promoter with the gene Neighbor of BRCA1 Number 1
(NBR1), which encodes autophagic receptor, reportedly having a role
in maintenance of cell stemness [38-40].
The choice of transcription start sites at human BRCA1/NBR2
promoter appears to have crucial importance for proper response of
the BRCA1 gene to various micro-environmental stimuli, including
genotoxic agents, DNA damage, estrogen stimulation and hypoxia.
However, so far reported studies have focused more on cancer or
transformed cell lines.
Hypoxia, pro-mitogenic activity of estrogens and DNA damage all
modulate the cellular redox (NAD/NADH) ratio. As found by [41],
an increased redox ratio uni-directionally enhances transcription from
the BRCA1 proximal promoter (Figure 1). This occurs through the
removal of co-repressors, including CtBP protein, (metabolic sensor of NAD/NADH ratio), BRCA1, HDAC from the promoter region. In
turn, unidirectional transcription of NBR2 lncRNA can be initiated
in response to energy stress (glucose starvation), by AMP-activated
protein (AMP) kinase, a key sensor of cellular energy status, (Figure
1). NBR2 lncRNA was observed to interact with activated AMPK
kinase and it is supposed to amplify and preserve AMPK activity
during chronic stress, which results in the repression of anabolic
processes of mTORC1 pathway, autophagy promotion, reduction of
cell proliferation) [42].
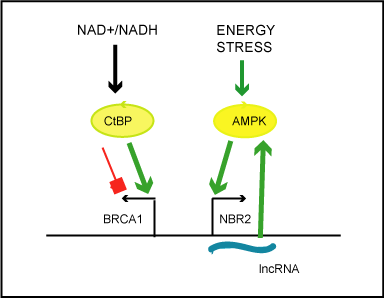
Figure 1. Simplified scheme illustrating the impact of the cellular metabolic states on transcriptional activity of the bidirectional BRCA1 promoter.
According to Di and colleagues, the redox ratio (NAD+/NADH) modulates the activity of
the metabolic sensor and transcriptional co-repressor, carboxyl-terminal binding protein
(CtBP).Increased NAD+/NADH ratio cause discharge of CtBP, from the BRCA1 promoter
and unidirectional enhancement of transcription from BRCA1 transcription start site.
Liu and colleagues state that energy stress (induced by glucose starvation) activates cellular
energy sensor, AMP-activated protein kinase, which by unknown mechanism induces
unidirectional transcription of lncRNA from NBR2 transcription start site. LncNBR2 has
been found to potentiate activity of AMPK under prolonged stress.
Interestingly, hypoxic stress decreases BRCA1 transcriptional
activity not only by modulation of redox ratio but also by dynamic
redistributions of E2Fs and pocket proteins at the BRCA1 promoter.
BRCA1 expression, which shows cell-cycle related pattern [43,44] is
E2F dependent. During normoxia, the two adjacent, conserved E2F
sites at the BRCA1 proximal promoter, within intergenic region,
are simultaneously occupied by E2F1 transcriptional activator
and E2F4/p130 transcriptional repressor. Hypoxia causes p130
dephosphorylation, an increase of the binding of repressive complex
E2F4/p130 to the intergenic region and the unidirectional repression of
BRCA1 transcription [45].
Extended hypoxia also induces repressive histone modification
changes, including decreased H3K4 methylation and leads to persistent
epigenetic silencing of the BRCA1 promoter [32].
In turn, oxidative stress stimulates BRCA1 transcription by
binding of an activated NRF2 transcription factor (Nuclear factorerythroid-
2p45-related factor 2), the master of redox switch, involved
in the Keap1-Nrf2-ARE pathway, to ARE sites (antioxidant response
elements) at the proximal BRCA1 promoter [46].
Estrogen has been found stimulate BRCA1 transcription either
by non-genomic mechanism (by changes in redox ratio, activation
of MAPK cascade) or potentially by recruitment of ER alfa to the
downstream BRCA1 promoter [47]. ER-alfa dependent activation
can be modulated by an aromatic hydrocarbon receptor complex,
which binds two consecutive xenobiotic-responsive elements located
upstream of the ER-alfa binding region [48].
The regulation of BRCA1 transcription is further influenced by a
number of other transcriptional factors, including, CREB [49], BP53
[50], c-Myc [51].
The bidirectional BRCA1/NBR2 promoter is bound by architectural
protein, CTCF transcription factor. The binding protects the promoter
region against DNA methylation, maintains its accessibility for
transcription factors and is critical for its functionality [52-54].
Mechanism of auto-regulation through co-residence of BRCA1,
E2F and Rb1 at the BRCA1 promoter has been also proposed by [55].
According to the authors, BRCA1 transcription is repressed by
BRCA1 and upregulated in response to genotoxic stress occurring after
the disruption of co-repressors array and dismissal of BRCA1 protein.
Subjecting the RB1 gene to genomic imprinting, that favor expression
from maternal allele [56] adds another layer to complexity of auto
regulation of BRCA1 transcription.
BRCA1 protein and its functions
BRCA1 protein
BRCA1 reportedly interacts with more than 100 of proteins and
has been proposed to act as a scaffold for the assembly of different
functional complexes, [57,58].
The BRCA1 C-terminal region contains two BRCT repeats, which
constitute a phospho-peptide binding domain, contributing to most of
BRCA1 functional interactions, including interactions with signaling
kinases ATM, ATR and CHK2. It can be transcriptionally active when
ligated with DNA binding domain.
The N-terminal RING domain (with its heterodimeric binding
partner, the BRCA1-associated RING-domain protein, BARD1)
displays an ubiquitin/ligase activity and functions as a highly active E3
Ub ligase in complexes with E2 ubiquitin (Ub)-conjugating enzymes.
The central, large region (60%) of BRCA1 acts as a scaffold and
interacts directly with DNA and proteins. It is required for homologous
recombination (HR) and checkpoint functions. Interestingly, it
preferentially binds to G-quadruplexes and other non-B-DNA
topologically constrained structures, which occure on numerous
promoter regions (e.g. C-Myc, KRAS, Kit, TERT genes) and on
telomeres [59].
BRCA1 functions
BRCA1 plays a critical role in multiple cellular processes required
for genome stability and cellular homeostasis. However, the mechanism
of how BRCA1 protein is responsible for increased risk of a breast
cancer is not fully understood.
BRCA1 in DNA damage response
The BRCA1 protein functions in a cellular DNA damage response
(DDR) network, responding to genotoxic stress. The network detects,
signals, repairs DNA/chromatin damage. It also coordinates the repair
process with cell cycle progression and cellular metabolism or directs
cells to apoptosis. DSBs may occur in result of DNA replication-errors,
ionizing radiation and oxidative stress [60]. However, they may also
be caused by programmed DSBs arising at specific locations in the
genome during meiosis as well as during V(D)J and immunoglobulin
heavy chain class switch recombination (CSR) [61]. Among DNA
lesions double strand breaks (DBS) are the most harmful as they may
induce severe detriment in DNA and chromatin organization and
cause chromosomal translocations. The selection process between the
two mechanisms for repairing DBS (error-prone non-homologous
end-joining [NHEJ] and homologous recombination [HR]) depends
on BRCA1 and on multiple factors, including DNA damage
checkpoints, ubiquitination steps and post-translational histone
modifications reviewed by [62]. In summary, a master sensor of DBS,
Ataxia-Telangiectasia Mutated (ATM) kinase induces histone H2AX
phosphorylation cascade and then a process of multi-steps recruitment
and assembly of damage signaling and repair factors. It also drives
chromatin modifications. The BRCA1 protein once phosphorylated
by ATM kinase counteracts inhibitory effect of chromatin barrier,
imposed on damage sites by BP53 and then initiates HR by activation
of DNA resection. As recently demonstrated, BRCA1-BARD1 E3
Ub ligase causes the repositioning of BP53 over long distance by
promoting activity of chromatin remodeler SMARCAD1 (SWI/SNF
Matrix-Associated Actin-Dependent Regulator Of Chromatin [63].
As a mediator of ATM signaling, BRCA1 activates DNA damage
checkpoints (G2/M phase), reviewed in [64]. It is worth noting, the
evidence suggesting that DBS repair pathways are developmentally
regulated with HR being crucial in embryonal cells and NHEJ during
cell cycles of differentiated cells. DBS repair by HR in primary somatic cells appears to require BRCA1, whereas ATM kinase is dispensable as
stated by [65].
Other molecular processes regulated by BRCA1
BRCA1/BARD1 heterodimer controls microtubule nucleation in
spindle assembly and centrosome duplication during mitosis [66].
Apart from repair damage and cell cycle progression control,
BRCA1 has other role attributed to its tumor suppression activity. It is
supposed to exert global effects on heterochromatin integrity through
transcriptional repression of satellite RNA (through ubiquitylation of
histone H2A) [67]. Recent reports provide evidence that BRCA1 (in
repressive complex with HP1 and DNMT3) may also cause global
heterochromatin silencing through ATM dependent DNA methylation
[68]. At heterochromatin regions, BRCA1 reportedly participate
in protection of DNA replication and it is required for HR at stalled
replication forks [69].
Furthermore protein has been found to function as a negative
regulator of Polycomb- repressive complex 2 (PCR2), which is
important for the maintenance of stem cell pluripotency and
suppression of cell differentiation [70].
Its role in transcriptional regulation is complex. BRCA1 regulates
transcription by association with basal transcriptional machinery
(Polymerase II and Polymerase I holoenzymes) and by interacting and
modulating the activity of numerous transcription factors (including
p53, c-myc, STAT1, E2F, NF-kB, OCT-1, estrogen, progesterone and
androgen receptors), transcriptional co-repressor and co-activators,
(including CtBP, Rb- and Rb-associated proteins, HDAC1/2, and p300,
HAT), chromatin remodeling complexes (specifically with BRG1-
central catalytic ATPase of ATP-dependent chromatin remodeling
complexes SWI/SNF [64].
Interestingly BRCA1 has been found at nuclear sub-compartments
with transcription machinery (transcriptional factories), that cluster
transcriptionally active or inactive genes [71]. Apart from protection
against genotoxic stress, DNA repair proteins at transcription factories
are also supposed to control programmed double strand breaks induced
by Topoisomerase 2 alfa for proper transcriptional output [72].
Throughout the genome BRCA1 resides at a large number of
gene promoters and regulates expression of specific subset of genes
in response to genotoxic stress or DNA damage [73,74]. BRCA1
transcriptional complexes regulate activity of pro- and anti-apoptotic
genes, genes involved in growth promotion, cell cycle arrest, DNA
repair, telomerase and interferon genes described by [58].
Its role in cellular metabolic homeostasis and reprogramming is
not completely understood.
BRCA1 is an important negative regulator of anabolic processes
promoted by estrogen receptor (ER) and functions in a negative
feedback loop, (activating transcription of ER and disrupts estrogen-
ER complex) reviewed by [75]. Moreover, it modulates IGF1/PIK3/Akt
pathway, (by transcriptional regulation of IGF1 as well as interaction
with AKT), [76,77] and fatty acid synthesis (maintaining acetyl-CoAcarboxylase
in an inactive state) [78]. BRCA1 has been also observed to
regulate NRF2 dependent antioxidant signaling and hypoxia response
[binding and stabilizing NRF2 transcription factor [79] and hypoxiainducible
factor-1α [HIF-1α] [80], respectively].
Recent metabolomics and transcriptomic data further suggests that
BRCA1 can cause reversion of aerobic glycolysis (known as a Warburg
effect) in breast cancer cells [81].
Long-range interactions and regulation of genome functions
Genome integrity is accomplished through regulation of DNA
replication and genes expression in the three dimensional nuclear
spaces.
Genomic long-range interactions (>10kb) are integral for 3D
genome organization. As proposed by [82], they can moderate
of communication along chromosome (in cis-) and between
chromosomes (in trans-). Bringing distant regulatory elements
(promoters, enhancers) into spatial proximity allows for effective
control of gene expression.
Genomic contacts are established partially by architectural
proteins (with key role of CTCF insulator protein and cohesin) and
are accompanied by looping out the intervening sequence, [83]. The
interactions may be influenced by various chromatin features, protein
cofactors and complexes, Mediator, DNA methylation and local
RNA transcription reviewed in [84]. Interestingly, basal transcription
machinery is known to be recruited by CTCF and cohesin [83,85].
Long-range interactions, detected by Hi-C on chromosomes
during interphase are observed as dynamically formed neighborhoods
(encompassing app. 400-500 MB), referred to as Topologically
Associated Domains (TADs), (Figure 2) [86,87]. The hierarchical
structure of inter-TADs contacts is established and mediated by several
factors, including architectural proteins, Mediator, tissue specific
transcription factors and local RNA transcription [88]. Interactions
within TADs correlate with expression levels and variability of
chromatin states [89,90]. Transcriptionally non-active, pre-existing
interactions are also observed. Jin and colleagues provided evidence,
that TNF-responsive gene promoters can be juxtaposed to TNFresponsive
regulatory elements prior to stimulation [91]. The authors
also suggest that once interactions are established in given cell they
might influence target gene expression in cell specific manner.
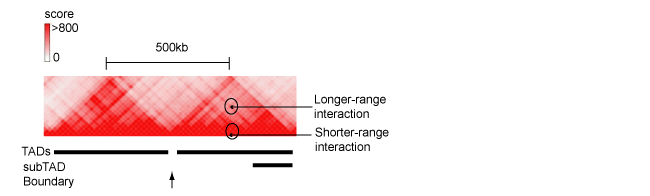
Figure 2. The fragment of Hi-C contacts map for chromosome 8 (http://www.3dgenome.org), demonstrating hierarchical structure of TAD. Positions of TAD, sub-TAD, domain boundary as well as long- and shorter-range interactions between genomic regulatory elements are indicated.
Epigenetic signatures of specialized TADs have been found to
correlate with hormones induced gene regulation [92]. Furthermore,
TADs reportedly align with DNA replication domains and were
proposed to represent stable units of replication-timing regulation [93].
It is worth noting, that spreading of histone H2AX phosphorylation,
induced by ATM kinase in response to DNA damage was detected along TADs. In contrast to H2AX, ATM kinase has been found locally,
on domains borders [94]2021 Copyright OAT. All rights reservggested,
that the major role of ATM kinase in DNA damage repair may rely
on its ability to modify both local as well as global chromosome
organization and chromatin mobility. It is presumed that this occurs
with contribution of actin filaments, microtubules and cohesin
complexes.
Although sequences within TADs interact preferentially with sites
inside the domain, at the edge, inter-domain and inter-chromosomal
contacts occur [95]. Borders domains, which separate TADs are
enriched in architectural proteins including CTCF and cohesin as
well as short interspersed nuclear elements (SINE) and tRNA genes
[93]. Their strength can be regulated developmentally, as in case of
the border, controlling interactions between HOXD genes and their
regulatory elements during mouse limb development [96]. The border
strength can also decrease in response to heat shock stress. Heat shock
in Drosophila was reported to cause an increase of inter-domain
and inter-chromosomal interactions between polycomb responsive
elements and the subsequent transcriptional silencing of entire TADs
domains [97]. Several reports and recent reviews describe the role of
local TAD boundary disruption in establishing improper regulatory
circuits (between oncogenes and regulatory elements) that can drive
neoplastic growth, discussed by [98,99].
As reported by Naumova and colleagues [100], interphase-specific
chromosomal organization of TADs is lost in mitotic cells and replaced
by a series of cell-invariant, consecutive loops. Dilep and colleagues
proposed that TADs and their long-range contacts are restablished
during early G1cell cycle phase coinciding with the establishment
of the replication-timing program [101]. According to Naumova
and colleaques higher order chromatin structures that have to form
de novo in early G1 do not themselves convey epigenetic memory
[100]. Instead, their re-emergence in early G1 is restored by histone
marks, DNA methylation, and protein complexes that remain on DNA
through mitosis, e.g. at key gene regulatory elements [102] or at TAD
boundaries [103]. In addition to polycomb group protein [102], tissuespecific
transcription factors [103] a possible role of Drosophila CTCF
in mitotic bookmarking and maintaining chromatin domains during
the cell cycle has been also suggested [104].
In this context it is worth to mention the recent evidence resulted
from mapping long-range genomic interactions before and after
reprogramming of somatic cells, that have demonstrated that specific
long-range contacts are acquired by induced pluripotent cells in celltype
specific-manner during reprogramming [105,106]. According to
Gonzales and Ng this indicates existence of topological memory in
reprogrammed somatic cells [107].
Long-range contacts were also mapped and analyzed on interphase
chromosomes, with close to single regulatory element resolution, by
Capture Hi-C (Chi-C), which combines Hi-C methodology with
hybridization-based capture of targeted genomic regions. These
analyses conducted along with Pol II precipitated interactions (by
ChiA-PET), revealed that both active and inactive genes promoters
contact each other and form multigenic complexes with correlated
expression levels. No bias was detected for active versus non-active
promoters [108,109]. According to Rowley and Corces this might
indicate the existence of so called “matrix of expression regulation”
[98]. Noteworthy, this evidence is also consistent with phenomenon
of clustering of co-regulated active and inactive genes observed at
nuclear sub-compartments, such as transcriptional factories discussed
by [72], Li and colleagues suggest, that clustering of gene promoters
can multiply an effect of any genetic error and/or polymorphism at
the single promoter level, depending on the cell specific factors [110].
Their evidence shows that the disease related SNPs are more likely to be
found at interacting promoter regions.
Long range interactions of the BRCA bidirectional promoter
To date dynamic long distance interactions between the promoter,
introns and terminator regions of the mammalian BRCA1 gene have
been reported [111]. The BRCA1 promoter and terminator contacts
have been found to suppress estrogen-induced transcription and be
potentially linked to dysregulated expression of BRCA1 seen in breast
tumors.
Publically available Hi-C contacts maps for chromosome 17,
localize the BRCA1/NBR2 promoter region app. 160 kb away from the
border of the TADs domain (in esc and IMR90 cells) (Figure S1[A])
(http://www.3dgenome.org/). Interestingly, this border, demonstrates
multiple inter-domain and inter-chromosomal interactions with X
chromosomes and autosomes in MCF-7 cells (not shown), (NIH
Roadmap Epigenomics Consortium; .
org/). Among several tools to analyze or to visualize Hi-C data, reviewed
recently by [112,113], Hi-C browser, by Ren lab, providesvirtual-4C
software, that supplements Hi-C data with DHS linkage and CHiPSeq
evidence (http://www.3dgenome.org/). The software visualized
cis- regulatory potential of SNP rs11655505 (C/T), at the bi-directional
BRCA1 promoter (Figure S1[B]) to establish long-range (>10kb)
contacts with regulatory elements at the Neighbor of BRCA1 number 1
(NBR1) gene, with the edge of TAD and with domain boundary (Figure
S2A[1,2,3]).
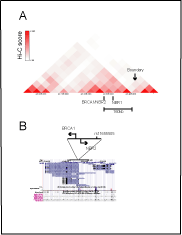
Figure S1. Interactions of the BRCA1 promoter at the Hi-C map for chromosome 17.
Position of the genetic variant (rs11655505) at the bi-directional BRCA1 promoter. Panel
A. Interactions of the BRCA1 bi-directional promoter are mapped app. 160 kb from the
border of the TADs domain in esc (http://genome.ucsc.edu/). Panel B. Polymorphic variant
(rs11655505) resides at the bi-directional BRCA1/NBR2 promoter and falls into region of
homology to the NBR1 regulatory elements. Position of SNP variant is referred to Ref-seq,
DNAse hypersensitivity sites (DHS), CTCF binding sites data, (http://genome.ucsc.edu/).
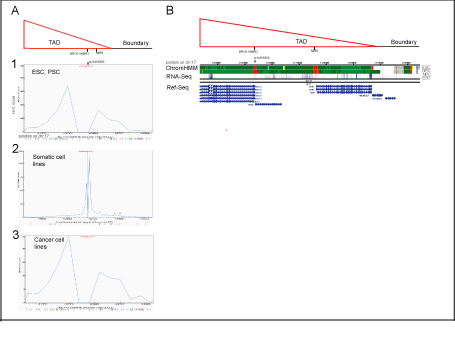
Figure S2: Potential cis-regulatory elements for SNP rs11655505 (C/T), and their chromatin states defined by virtual-4C and chromHMM analysis, respectively.
Panel A
Linear plots of rs11655505 quantified interactions obtained by 4-C virtual analysis (http://www.3dgenome.org,/a>), which simulates Hi-C data,
supplements it with DHS linkage and CHIP-Seq data, aligned with depicted TAD and the boundary (proximal to BRCA1 locus).
A[1]
In embrional stem cells and progenitor cells (including H1-ESC, H1-MSC, H1-NPC), rs11655505 demonstrates potential to contact the NBR1 gene regulatory element as well as the TADs
edge and TADs boundary. The interacting potential of rs11655505 was visualized by Hi-C browser with resolution of 40 kb.
A[2]
In somatic cell lines (including HMEC, HUVEC, NHEK, IMR90, GM12878), rs11655505 shows the highest potential for short-range interactions (<10kb) (within the NBR2 sequences),
defined by the software with resolution of 5 kb.
A[3]
In cancer cell lines (including, PANCI, LNCaP, Caki2), patterns of potential long-range interacting elements for rs11655505, (predicted by the software with resolution of 40 kb), are similar
to these shown in esc and psc.
Panel B
In esc and psc, potential long-range interacting elements at the BRCA1, NBR2 and NBR1 genes are characterized as the active promoters, by chromHMM.
In these cells, the transcripts of BRCA1, NBR2 and NBR1 are detected by RNA-seq (NIH Roadmap Epigenomics Consortium; http://www.roadmapepigenomics.org/).
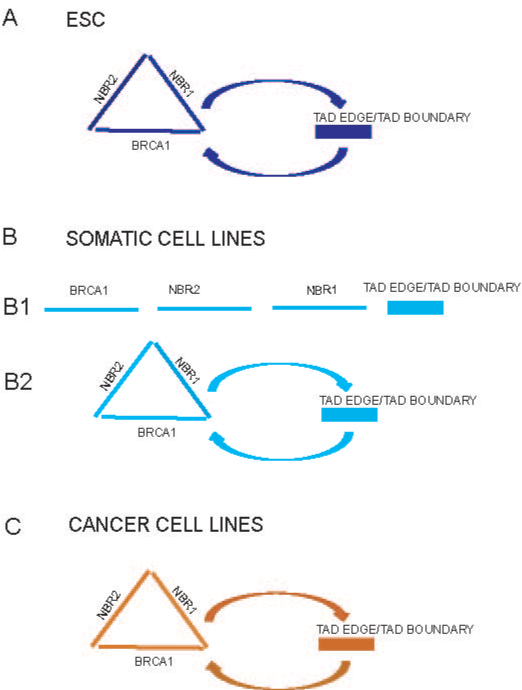
Figure 3. The outline of potential long-range interactions of the BRCA1 promoter (driven
by polymorphic variant SNP rs11655505 C>T, as an example).
In embryonal stem cells, the BRCA1, NBR2 and NBR1 active promoters may cluster
together and/or establish long-range interactions with TADs edge and TADs boundary
(Figure 3[A]).
In somatic and cancer cell lines, the active BRCA1, NBR2, NBR1 promoters and/or
enhancers potentially interact and contact with TADs edge and TADs boundary (Figure
3B[2], C).
One may speculate, that these potential long–range interactions might increase
communication between the BRCA1, NBR2, NBR1 regulatory elements and TADs edge
or boundary in cell specific manners, (marked by different colors), (Figure 3A, B[2], C).
The vast majority of cells from somatic cell lines have low potential to make the long-range
contacts with regulatory elements of the NBR1 gene and TADs boundary (Figure 3B[1]).
Based on visualization of genomic contacts of rs11655505 by 4C-virtual software (http://www.3dgenome.org/), as well as data from public resources, provided by (NIH Roadmap
Epigenomics Consortium; http://www.roadmapepigenomics.org/)
In H1 embryonic stem cells long-range interacting regulatory
elements of the BRCA1, NBR2, NBR1 genes are characterized as an
active promoters (Figure S2B). Whereas in somatic and in cancer
cell lines they are defined as active promoters and/or enhancers (not
shown) (ChromHMM; NIH Roadmap Epigenomics Consortium; http://www.roadmapepigenomics.org/). Their evolutionary profiles
show the presence of subregions with over 98% of homology resulted
from segmental duplication. Moreover the BRCA1, NBR1 genes and
lncRNA of NBR2 are significantly expressed in H1 embryonic stem
cells (RNA-Seq data Roadmap Epigenomics Consortium; http://www.roadmapepigenomics.org/) (Figure S2[B]).
As the whole, the publicly available data allow to speculate, that the
BRCA1, NBR2 and NBR1 promoters may cluster together and have correlated transcription level (which might be referred as a putative
so called “matrix of expression regulation” in embryonic stem cells,
(Figure 3A). Potential interactions between promoters are of special
interests because their clustering might multiply the risk of the single
genetic variant (SNPs).
Furthermore, the potential contacts of the BRCA1 promoter with
TADs edge and/or boundary domain might enable transmission cellautonomous
signals through the inter-domain and inter-chromosomal
contacts (Figure 3[A]). One may speculate that these signals could be
related to sexual identity and/or parental of origin effects. Notably,
sexual identity of adult stem cells with XX karyotypes has recently
been reported to have a novel significant role in controlling organ size,
plasticity and tumor susceptibility (in Drosophila intestine) [114]. The
potential interactions might also connect the BRCA1 promoter and its
transcriptional machinery, (bearing autoregulatory BRCA1 protein)
with ATM kinase (if it is accumulated at the boundary) and modify the
risk of BRCA1 mutations.
The establishment of long-range genomic contacts (mediated
by numerous nuclear factors) is never a one-sided effect and it may influence status of TAD’s edge and its border domain. In that case the
putative BRCA1 promoter regulatory circuit, (displayed by rs11655505)
might have an impact on chromatin status and expression rate of the
BRCA1 promoter, many other genes in cis- and in trans- and contribute
to the potential epigenetic memory of the region.
In contrast to H1 embryonal stem cells, in somatic and cancer cell
lines, the putative long-range interactions may be driven by the BRCA1,
NBR2, NBR1 active promoters and/or enhancer regulatory elements
(Figure 3B[2], 3C). However, based on virtual 4-C analysis, one may
speculate that the vast majority of cells in somatic cell lines does not
establish long-range contacts between the BRCA1/NBR2, NBR1
regulatory elements and TADs boundary (Figure 3B[1]).
Conclusions
In each cell BRCA1 mediates molecular processes, which are
critical for genome stability. In mammary gland BRCA1 expression
is required for differentiation of breast stem cells and its disturbance
may be implicated in aetiology of basal-like breast cancer. Longrange
contacts of the bidirectional BRCA1 promoter with the NBR1
regulatory elements, TADs edge and TADs boundary are of interest
because they might moderate the BRCA1 promoter communication
along chromosome as well as between chromosome and increase
BRCA1 expression plasticity in response to genotoxic stress. Once
established they could be implicated in maintenance of pluripotency
and contribute to potential epigenetic memory of region. They might
also multiply the risk of the genetic variants at the BRCA1 promoter
due to clustering of promoters (associated with establishment of
putative so called “matrix of expression regulation”) or to modify the
risk of mutations of the BRCA1 protein. Comparative, high resolution
mapping and analysis of long-range contacts of the BRCA1 promoter in
stem, progenitors, primary somatic and cancer cells, as well as in breast
cancer patients, when it becomes attainable, will shed more light on
their potential contribution to the risk of breast cancer.
Acknowledgements
The author thanks Dr Ren and Dr Yue for providing the Hi-C genome browser as well as the NIH Roadmap Epigenomic Consortium for CHIP-Seq, RNA-seq and Hi-C datasets.
The research was supported by National Science Centre (NCN) grant 2013/11/B/NZ2/00132.
References
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, et al. (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136: E359-386. [Crossref]
- Welch DR (2016) Tumor Heterogeneity--A 'Contemporary Concept' Founded on Historical Insights and Predictions. Cancer Res 76: 4-6. [Crossref]
- Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS (2003) Stem cells in normal breast development and breast cancer. Cell Prolif 36 Suppl 1: 59-72. [Crossref]
- Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M (2010) Genetic susceptibility to breast cancer. Mol Oncol 4: 174-191. [Crossref]
- Collaborative Group on Hormonal Factors in Breast Cancer (2001) Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 358: 1389–1399. [Crossref]
- O'Brien KM, Shi M, Sandler DP, Taylor JA, Zaykin DV, et al. (2016) A family-based, genome-wide association study of young-onset breast cancer: inherited variants and maternally mediated effects. Eur J Hum Genet 24: 1316–1323. [Crossref]
- Weinberg CR, Shi M, DeRoo LA, Taylor JA, Sandler DP, et al. (2014) Asymmetry in family history implicates nonstandard genetic mechanisms: application to the genetics of breast cancer. PLoS Genet 10: e1004174. [Crossref]
- Milne RL, Antoniou AC (2016) Modifiers of breast and ovarian cancer risks for BRCA1 and BRCA2 mutation carriers. Endocr Relat Cancer 23: T69-84. [Crossref]
- Cox DG, Simard J, Sinnett D, Hamdi Y, Soucy P, et al. (2011) Common variants of the BRCA1 wild-type allele modify the risk of breast cancer in BRCA1 mutation carriers. Hum Mol Genet 20: 4732-4747. [Crossref]
- Mavaddat N, Pharoah PD, Michailidou K, Tyrer J, Brook MN, et al. (2015) Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst 107. [Crossref]
- Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, et al. (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447: 1087–1093. [Crossref]
- Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, et al. (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45: 353–361e2. [Crossref]
- Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, et al. (2015) Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet 47: 373–380. [Crossref]
- Meyer KB, O’Reilly M, Michailidou K, Carlebur S, Edwards SL, et al. (2013) Fine-Scale Mapping of the FGFR2 Breast Cancer Risk Locus: Putative Functional Variants Differentially Bind FOXA1 and E2F1. Am J Hum Genet 93: 1046–1060. [Crossref]
- Wang X, Tucker NR, Rizki G, Mills R, Krijger PH, et al. (2016) Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. eLife 5: e10557. [Crossref]
- Schierding W, Cutfield WS, O’Sullivan JM (2014) The missing story behind Genome Wide Association Studies: single nucleotide polymorphisms in gene deserts have a story to tell. Front Genet 5: 39. [Crossref]
- Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP (2004) The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst 96: 1659-1668. [Crossref]
- Golubnitschaja O, Debald M, Yeghiazaryan K, Kuhn W, Pešta M, et al. (2016) Breast cancer epidemic in the early twenty-first century: evaluation of risk factors, cumulative questionnaires and recommendations for preventive measures. Tumour Biol 37: 12941-12957. [Crossref]
- Romagnolo AP, Romagnolo DF, Selmin OI1 (2015) BRCA1 as target for breast cancer prevention and therapy. Anticancer Agents Med Chem 15: 4-14. [Crossref]
- Bitzur R, Brenner R, Maor E, Antebi M, Ziv-Baran T, et al. (2016) Metabolic syndrome, obesity, and the risk of cancer development. Eur J Intern Med 34: 89-93. [Crossref]
- Hauner D, Hauner H (2014) Metabolic syndrome and breast cancer: is there a link? Breast Care (Basel) 9: 277-281. [Crossref]
- Link JC, Chen X, Arnold AP, Reue K (2013) Metabolic impact of sex chromosomes. Adipocyte 2: 74-79. [Crossref]
- Lord CJ, Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16: 110–120. [Crossref]
- Wei M, Grushko TA, Dignam J, Hagos F, Nanda R, et al. (2005) BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res 65: 10692–10699. [Crossref]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, et al. (2008) An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40: 499–507. [Crossref]
- De Summa S, Pinto R, Sambiasi D, Petriella D, Paradiso V, et al. (2013) BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol 24 Suppl 8: viii13-13viii21. [Crossref]
- Turner NC, Reis-Filho JS (2006) Basal-like breast cancer and the BRCA1 phenotype. Oncogene 25: 5846-5853. [Crossref]
- Di Oto E, Monti V, Cucchi MC, Masetti R, Varga Z4, et al. (2015) X chromosome gain in male breast cancer. Hum Pathol 46: 1908-1912. [Crossref]
- Carone DM, Lawrence JB (2013) Heterochromatin instability in cancer: from the Barr body to satellites and the nuclear periphery. Semin Cancer Biol 23: 99-108. [Crossref]
- Horsman MR, Vaupel P (2016) Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front Oncol 6: 66. [Crossref]
- Yan M, Rayoo M, Takano EA; KConFab Investigators, Fox SB (2009) BRCA1 tumours correlate with a HIF-1a phenotype and have a poor prognosis through modulation of hydroxylase enzyme profile expression. Br J Cancer 101: 1168–1174. [Crossref]
- Lu Y, Chu A, Turker MS, Glazer PM (2011) Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol Cell Biol 31: 3339-3350. [Crossref]
- Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, et al. (2008) BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A 105: 1680-1685. [Crossref]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, et al. (2016) Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A 113: E2047-2056. [Crossref]
- Dimitrov S, Brennerova M, Forejt J (2001) Expression profiles and intergenic structure of head-to-head oriented Brca1 and Nbr1 genes. Gene 262: 89-98. [Crossref]
- Xu CF, Brown MA, Nicolai H, Chambers JA, Griffiths BL, et al. (1997) Isolation and characterisation of the NBR2 gene which lies head to head with the human BRCA1 gene. Hum Mol Genet 6: 1057-1062. [Crossref]
- Pavlicek A, Noskov VN, Kouprina N, Barrett JC, Jurka J, et al. (2004) Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum Mol Genet 13: 2737-2751. [Crossref]
- Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, et al. (2016) NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 212: 577-590. [Crossref]
- Kenific CM, Debnath J (2016) NBR1-dependent selective autophagy is required for efficient cell-matrix adhesion site disassembly. Autophagy 12: 1958-1959. [Crossref]
- Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, et al. (2011) Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 13: 1214–1223. [Crossref]
- Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K (2010) Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol 17: 1406-1413. [Crossref]
- Liu X, Xiao ZD, Gan B (2016) An lncRNA switch for AMPK activation. Cell Cycle 15: 1948-1949. [Crossref]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, et al. (1997) Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90: 425–435.
- Vaughn JP, Davis PL, Jarboe MD, Huper G, Evans AC, et al. (1996) BRCA1 expression is induced before DNA synthesis in both normal and tumor-derived breast cells. Cell Growth Differ 7: 711–715. [Crossref]
- Bindra RS, Glazer PM (2006) Basal repression of BRCA1 by multiple E2Fs and pocket proteins at adjacent E2F sites. Cancer Biol Ther 5: 1400-1407. [Crossref]
- Wang Q, Li J, Yang X, Sun H, Gao S, et al. (2013) Nrf2 is associated with the regulation of basal transcription activity of the BRCA1 gene. Acta Biochim Biophys Sin (Shanghai) 45: 179-187. [Crossref]
- Jeffy BD, Hockings JK, Kemp MQ, Morgan SS, Hager JA, et al. (2005) An estrogen receptor-alpha/p300 complex activates the BRCA-1 promoter at an AP-1 site that binds Jun/Fos transcription factors: repressive effects of p53 on BRCA-1 transcription. Neoplasia 7: 873–882. [Crossref]
- Hockings JK, Thorne PA, Kemp MQ, Morgan SS, Selmin O, et al. (2006) The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res 66: 2224–2232. [Crossref]
- Atlas E, Stramwasser M, Mueller CR (2001) A CREB site in the BRCA1 proximal promoter acts as a constitutive transcriptional element. Oncogene 20: 7110-7114. [Crossref]
- Rauch T, Zhong X, Pfeifer GP, Xu X (2005) 53BP1 is a positive regulator of the BRCA1 promoter. Cell Cycle 4: 1078-1083. [Crossref]
- Chen Y, Xu J, Borowicz S, Collins C, Huo D, et al. (2011) c-Myc activates BRCA1 gene expression through distal promoter elements in breast cancer cells. BMC Cancer 11: 246. [Crossref]
- Butcher DT, Mancini-DiNardo DN, Archer TK, Rodenhiser DI (2004) DNA binding sites for putative methylation boundaries in the unmethylated region of the BRCA1 promoter. Int J Cancer 111: 669–678.
- Butcher DT, Rodenhiser DI (2007) Epigenetic inactivation of BRCA1 is associated with aberrant expression of CTCF and DNA methyltransferase (DNMT3B) in some sporadic breast tumours. Eur J Cancer 43: 210-219. [Crossref]
- Xu J, Huo D, Chen Y, Nwachukwu C, Collins C, et al. (2010) CpG island methylation affects accessibility of the proximal BRCA1 promoter to transcription factors. Breast Cancer Res Treat 120: 593–601. [Crossref]
- De Siervi A, De Luca P, Byun JS, Di LJ, Fufa T, et al. (2010) Transcriptional autoregulation by BRCA1. Cancer Res 70: 532-542. [Crossref]
- Kanber D, Berulava T, Ammerpohl O, Mitter D, Richter J, et al. (2009) The human retinoblastoma gene is imprinted. PLoS Genet 5: e1000790. [Crossref]
- Jiang Q, Greenberg RA2 (2015) Deciphering the BRCA1 Tumor Suppressor Network. J Biol Chem 290: 17724-17732. [Crossref]
- Savage KI, Harkin DP (2015) BRCA1, a 'complex' protein involved in the maintenance of genomic stability. FEBS J 282: 630-646. [Crossref]
- Brázda V, Hároníková L, Liao JC, Fridrichová H, et al. (2016) Strong preference of BRCA1 protein to topologically constrained non-B DNA structures. BMC Mol Biol 17: 14. [Crossref]
- Mortusewicz O, Herr P, Helleday T (2013) Early replication fragile sites: where replication-transcription collisions cause genetic instability. EMBO J 32: 493–495. [Crossref]
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B (2013) Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152: 417-429. [Crossref]
- Daley JM, Sung P (2014) 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 34: 1380-1388. [Crossref]
- Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, et al. (2016) Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 23: 647–655. [Crossref]
- Rosen EM (2013) BRCA1 in the DNA damage response and at telomeres. Front Genet 4: 85. [Crossref]
- Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, et al. (2013) Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc Natl Acad Sci USA 110: 5564–5569. [Crossref]
- Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, et al. (2004) BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol 24: 8457-8466. [Crossref]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, et al. (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477: 179-184. [Crossref]
- Filipponi D, Muller J, Emelyanov A, Bulavin DV (2013) Wip1 controls global heterochromatin silencing via ATM/BRCA1-dependent DNA methylation. Cancer Cell 24: 528-541. [Crossref]
- Huh MS, Ivanochko D, Hashem LE, Curtin M, Delorme M, et al. (2016) Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis 7: e2220. [Crossref]
- Wang L, Zeng X, Chen S, Ding L, Zhong J, et al. (2013) BRCA1 is a negative modulator of the PRC2 complex. EMBO J 32: 1584-1597. [Crossref]
- Jackson DA (2005) The amazing complexity of transcription factories. Brief Funct Genomic Proteomic 4: 143-157. [Crossref]
- Osborne CS (2014) Molecular pathways: transcription factories and chromosomal translocations. Clin Cancer Res 2: 296–300. [Crossref]
- Gardini A, Baillat D, Cesaroni M, Shiekhattar R (2014) Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J 33: 890-905. [Crossref]
- Gorski JJ, Savage KI, Mulligan JM, McDade SS, Blayney JK, et al. (2011) Profiling of the BRCA1 transcriptome through microarray and ChIP-chip analysis. Nucleic Acids Res 39: 9536-9548. [Crossref]
- Wang L, Di LJ (2014) BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact? Int J Biol Sci 10: 566-575. [Crossref]
- Kang HJ, Yi YW, Kim HJ, Hong YB, Seong YS, et al. (2012) BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis 3: e336. [Crossref]
- Xiang T, Ohashi A, Huang Y, Pandita TK, Ludwig T, et al. (2008) Negative Regulation of AKT Activation by BRCA1. Cancer Res 68: 10040-10044. [Crossref]
- Magnard C, Bachelier R, Vincent A, Jaquinod M, Kieffer S, et al. (2002) BRCA1 interacts with acetyl-CoA carboxylase through its tandem of BRCT domains. Oncogene 21: 6729-6739. [Crossref]
- Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, et al. (2009) Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell 34: 663–673. [Crossref]
- Kang HJ, Kim HJ, Rih JK, Mattson TL, Kim KW, et al. (2006) BRCA1 plays a role in the hypoxic response by regulating HIF-1alpha stability and by modulating vascular endothelial growth factor expression. J Biol Chem 281: 13047–13056. [Crossref]
- Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, et al. (2014) BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PloS One 9: e102438. [Crossref]
- Dekker J, Mirny L (2016) The 3D Genome as Moderator of Chromosomal Communication. Cell 164: 1110-1121. [Crossref]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, et al. (2006) CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 20: 2349–2354. [Crossref]
- Cubeñas-Potts C, Corces VG (2015) Architectural proteins, transcription, and the three-dimensional organization of the genome. FEBS Lett 589: 2923–2930. [Crossref]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, et al. (2015) CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163: 1611–1627. [Crossref]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376-380. [Crossref]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, et al. (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [Crossref]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, et al. (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153: 1281–1295. [Crossref]
- Grubert F, Zaugg JB, Kasowski M, Ursu O, Spacek DV, et al. (2015) Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell 162: 1051–1065.
- Waszak SM, Delaneau O, Gschwind AR, Kilpinen H, Raghav SK, et al. (2015) Population Variation and Genetic Control of Modular Chromatin Architecture in Humans. Cell 162: 1039–1050.
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, et al. (2013) A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503: 290–294. [Crossref]
- Le Dily F, Baù D, Pohl A, Vicent G, Soronellas D, et al. (2014) Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev 28: 2151–2162. [Crossref]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, et al. (2014) Topologically associating domains are stable units of replication-timing regulation. Nature 515: 402–405. [Crossref]
- Aymard F, Legube G (2016) A TAD closer to ATM. Mol Cell Oncol 3: e1134411. [Crossref]
- Goloborodko A, Marko JF, Mirny LA (2016) Chromosome Compaction by Active Loop Extrusion. Biophys J 110: 2162-2168. [Crossref]
- Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, et al. (2013) A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340: 1234167. [Crossref]
- Li L, Lyu X, Hou C, Takenaka N, Nguyen HQ, et al. (2015) Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol Cell 58: 216–231.
- Corces MR, Corces VG (2016) The three-dimensional cancer genome. Curr Opin Genet Dev 36: 1-7. [Crossref]
- Valton AL, Dekker J (2016) TAD disruption as oncogenic driver. Curr Opin Genet Dev 36: 34-40. [Crossref]
- Naumova N1, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, et al. (2013) Organization of the mitotic chromosome. Science 342: 948-953. [Crossref]
- Dileep V, Ay F, Sima J, Vera DL, Noble WS, et al. (2015) Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res 25: 1104–1113. [Crossref]
- Follmer NE, Wani AH, Francis NJ (2012) A polycomb group protein is retained at specific sites on chromatin in mitosis. PLoS Genet 8: e1003135. [Crossref]
- Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, (2012) Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150: 725–737. [Crossref]
- Shen W, Wang D, Ye B, Shi M, et al. (2015) A possible role of Drosophila CTCF in mitotic bookmarking and maintaining chromatin domains during the cell cycle. Biol Res 48: 27. [Crossref]
- Beagan JA, Gilgenast TG, Kim J, Plona Z, Norton HK, et al. (2016) Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell Stem Cell 18: 611–624. [Crossref]
- Krijger PH, Di Stefano B, de Wit E, Limone F, van Oevelen C, et al. (2016) Cell-of-Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell 18: 597-610. [Crossref]
- Gonzales KA, Ng HH (2016) Looping around Reprogramming: The Topological Memory of Induced Pluripotency. Cell Stem Cell 18: 557-559. [Crossref]
- Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 47: 598–606. [Crossref]
- Sahlén P, Abdullayev I, Ramsköld D, Matskova L, Rilakovic N, et al. (2015) Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biol 16: 156. [Crossref]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, et al. (2012) Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148: 84–98. [Crossref]
- Tan-Wong SM, French JD, Proudfoot NJ, Brown MA (2008) Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc Natl Acad Sci USA 105: 5160ndash;5165.[Crossref]
- Denker A, de Laat W (2016) The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev 30: 1357-1382. [Crossref]
- Mora A, Sandve GK, Gabrielsen OS, Eskeland R (2016) In the loop: promoter-enhancer interactions and bioinformatics. Brief Bioinform 17: 980-995. [Crossref]
- Hudry B, Khadayate S, Miguel-Aliaga I (2016) The sexual identity of adult intestinal stem cells controls organ size and plasticity. Nature 530: 344-348. [Crossref]