Abstract
Introduction: Neuropathy is an agonizing debility seen commonly in diabetic patients. Statins can also lead to similar neuropathic symptoms independent of diabetic status. When statins are prescribed in patients of diabetes, it is not possible to distinguish whether neuropathic pain is secondary to diabetes or to statin therapy. This review analyzes the molecular mechanisms involved in etio pathogenesis of neuropathy caused either by diabetes or by statin therapy. ATP deficiency and ion channel abnormalities may be responsible for the symptoms of neuropathy in diabetes, and a similar mechanism is at play in statin induced neuropathy. This is seen to occur independent of blood sugar levels.
Areas Covered: Literature published between 1980 and 2019 on the etio pathogenesis of neuropathy, insulin receptor defects, cholesterol biosynthesis, action of statins and of Coenzyme Q, KATP and calcium channels was reviewed. A comprehensive search on PubMed, Embase and Cochrane databases was carried out.
Expert Opinion: Statins activate KATP channels and directly affect neurons. Activation of KATP channels inhibits calcium channels which should theoretically be beneficial in neuropathic symptoms. However, statins lead to a simultaneous deficiency in levels of ubiquinone because of their inhibitory action on cholesterol biosynthesis pathway, This leads to neuropathy. Insulin receptor defects in diabetes may be leading to a similar pathogenesis in the causation of diabetic neuropathy.
Introduction
Statins are prescribed widely for both primary and secondary prevention of coronary artery disease. They act by their lipid lowering action, as well as by their pleiotropic effects. The number of patients who are on statin therapy is gradually and steadily increasing [1,2] Statins have been found to be generally safe; but they may be responsible for the side effects which so far have been attributed to the disease process per se for which they are being prescribed; and long term trials specifically exploring this aspect would unravel their real safety [3,4]
That statins lead to increasing incidence of new onset diabetes has recently been reported in literature [5]. Since a large number of patients who have been prescribed statins are already suffering from diabetes, it is practically not possible to evaluate whether the worsening of diabetes in these patients is because of diabetes per se or because of statin therapy.
Statin therapy per se may be causing polyneuropathy, which itself is a common complication of diabetes. It is difficult to interpret whether the reported polyneuropathy is because of statin therapy in a diabetic patient, or because of disease process of diabetes per se. This becomes significant because statins are prescribed very commonly in diabetic patients.
In a nested case control study [6] in country of Funen comprising 465,000 inhabitants, ‘first time ever’ cases of polyneuropathy registered in between 1994 and 1998 were identified. Using relevant statistical analysis and a prescription register to assess drug exposure; the researchers estimated the odds ratio of statin use in case of polyneuropathy as compared to control subjects, and concluded that chronic use of statins increased the risk of polyneuropathy significantly. There was a 4-14 fold increased risk of developing polyneuropathy as compared to general population.
Statin use and peripheral neuropathy has been found to be interlinked in many studies [7-11]. A 36 months prospective clinical and neurophysiological follow up in 42 patients with dyslipidaemia was conducted where patients were initiated on statin therapy ie Simvastatin 20mg daily and followed up. This study confirmed that statins cause a silent but definite damage to peripheral nerves if the treatment is given for more than 2 years [12]. A recent study suggests that statin consumption can lead to axonal involvement preferentially while causing damage to the peripheral nerves [13].
Though statins are used in all diabetic patients irrespective of lipid levels due to their pleiotropic effects, there use is almost universal in patients of diabetes with dyslipidemia. Whether neuropathy which manifests in these diabetic patients is because of statin therapy or because of diabetes has not been discussed in literature so far.
Present hypothesis is that disprortionately high lipid levels are responsible for neuropathy in dyslipidemic patients, and hyperlipidemia is one of the contributing factors in the etiology of neuropathy. However, axonal sensory motor large nerve fiber involvement is the one seen in patients suffering from neuropathy on statin therapy; in contrast to the one seen in patients with hyperlipidemia induced neuropathy, where pain symptoms consistent with small nerve fibre neuropathy are seen [14-19].
Statins inhibit the synthesis of ubiquinone. We propose in this review that the ubiquinone deficiency caused due to statin therapy may be one of the factors leading to neuropathy in patients who are being administered these medications, How decreased production of ubiquinone in patients on statin therapy is responsible for neuropathy has been discussed in this review. This review will focus on the actions of statins on body tissues at the cellular level; and the molecular mechanisms involved in HMG CoA reductase inhibition The pleiotropic actions of statins shall also be discussed in brief.
Material and Methods
A comprehensive search has been carried out on PubMed, Cochrane and Medline Embase and the molecular mechanisms, available in literature between 1980 to 2019, involved in neuropathy and statin induced neuropathy have been analyzed and discussed. Role of calcium channels and that of ubiquinone has been discussed in relation to polyneuropathy. The review concludes by strengthening the premise built thereon that statins lead to polyneuropathy primarily by decreasing the production of ubiquinone.
- Polyneuropathy is a known complication of diabetes. Statins prescribed commonly in diabetes may also cause similar symptomatology.
- In diabetes, mitochondrial function and cellular calcium levels are altered by impaired signals directed by defects in insulin receptors, independent of hyperglycemia.
- Decreased stimulation of insulin receptors leads to mitochondrial dysfunction which leads to decreased ATP production.
- Decreased ATP levels cause malfunctioning of plasmalemmal and ER calcium pumps.
- Decreased ER calcium uptake results in decreased calcium concentration in intra ER compartment. This results in decreased supply of voltage gated channels.
- Statins, by inhibiting cholesterol biosynthesis pathway, cause decreased ATP production, KATP channel activation, and thereby inhibition of VGCC.
- Neuropathy caused by diabetes and statins occurs by similar pathophysiological mechanisms. Statin administration increases neuropathy incidence in diabetes and has additive symptomatic effect.
- Ubiquinone increases ATP levels and its supplementation to statin therapy seems to have a rationale basis in alleviating the ailment of polyneuropathy.
|
Sensory neuropathy
Histology of the neural tissue exhibiting Neuropathy
In diabetic neuropathy, histological examination of the neural tissue reveals microangiopathy; involvement of schwan cells, degeneration of axons and axonal demylenation [8-10]. Maximal neuronal degeneration is observed in the longest axons of sensory neurons [18].
Oxidative stress, involvement of polyol pathway, elevated reactive oxygen species, and advanced glycated end products have all been implicated in the pathophysiology of diabetic neuropathy [18,19]
Pathogenesis of sensory neuropathy and the role of ion channels
Pain sensation is initiated or sustained by neural afferent overactivity. This occurs when peripheral neuronal activity is increased, or when there is a mismatch between DRG cell bodies and adjacent neuron cross excitation. Axonal firing is determined by voltage gated sodium channels. Local anesthetics are exploited to alleviate pain because they block these voltage gated sodium channels [20].
Changes in intracellular calcium levels also determine sensory neuron excitability[21-23] Cav 3.2 subtype calcium channels (low voltage activated T-type calcium channels) located on primary afferent terminals and cell bodies are exploited for modulating neuronal excitability. A decrease in expression of these channels results in alleviation of neuropathic pain. Elevated calcium concentration in cytoplasm secondary to chronic dysregulation of calcium homeostasis leads to increased aberrant pain sensation [24].
Diabetes tends to involve sensory pathways in a symmetrical manner and the nerve conduction studies in the affected patients suggest a diminishing nerve conduction velocity.
Pathogenesis of sensory neuropathy
Diabetic neuropathy: and the role of ATP and ion channels
[25] One of the proposed mechanisms in the etiopathogenesis of diabetic neuropathy is that the mitochondrial function and cellular calcium levels are altered; and this is caused not by hyperglycemia per se in a diabetic patient, but by impaired signals directed by changes in insulin receptors. A decreased stimulation of insulin receptors leads to dysfunction of mitochondria, and this dysfunction leads to decreased ATP production.
A decreased level of ATP alters the functioning of plasmalemmal and ER calcium pumps. ER calcium uptake is lowered and this results in decreased calcium concentration in intra ER compartment. This leads to decreased supply of voltage gated channels. Nerve conduction velocity is thereby diminished. When this is coupled later on with other pathways associated with hyperglycemia viz. polyol pathway, oxidative stress, protein glycosylation, the result is manifestation neuropathic symptoms.
Diabetes can lead to polyneuropathy by at least three mechanisms. They are
- calcium ion channel dysregulation
- decreased ATP production.
- activation of KATP channels
It is interesting to note that administration of statins in patients leads to both disturbances in calcium ion channel regulation, and also decreased synthesis of ATP, irrespective of their diabetic status.
Statin induced neuropathy: statins lead to calcium ion deregulation, and decrease in ATP production
Statins are inhibitors of an enzyme called 3 hydroxy methylglutoryl coenzyme A (HMG CoA) reductase. HMG CoA reductase reduces HMG CoA to mevalonate. HMG CoA reductase enzyme is regulated by cholesterol supply and is a rate limiting enzyme in cholesterol biosynthesis pathway. Statins are competitive inhibitors of HMG CoA reductase. Mevalonate is converted to squalene by a series of enzymatic reactions. Squalene epoxidase and oxido squalene cyclase act on squalene thus formed and convert it to lanosterol. A cascade of chemical reactions then leads to formation of cholesterol from lanosterol (Figure 1).
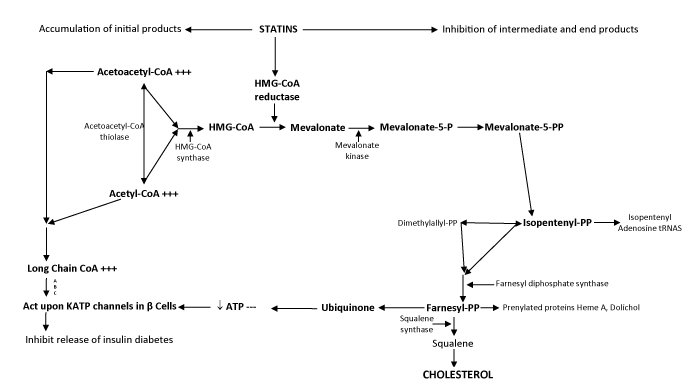
Figure 1: The cholesterol biosynthesis pathway. Some of the major intermediates and end -products are depicted. Enzymes in the pathway are found in cystosol, endoplasmic reticulum (ER) and peroxisomes, as noted. Figure adapted from Olivier and Krisans60, 3-hydroxy-3-methylglutaryl; DHC, dehydrocholesterol.
Olivier LM, Krisans SK. Peroxisomal protein targeting and identification of peroxisomal targeting signals in cholesterol biosynthetic enzymes. Biochim Biophys Acta, 2000;1529(1-3): 89-102.
Besides reduction in synthesis of cholesterol;prenylated proteins, heme A and dolichol generation is also reduced by inhibition of HMG CoA reductase. More importantly, ubiquinone production is decreased. Ubiquinone is an important component of electron transport chain. It participates in aerobic cellular respiration and generates energy in form of ATP. As is evident from figure 1, inhibition of cholesterol biosynthesis pathway by HMG CoA reductase inhibitors leads to decreased production of cholesterol, ubiquinone and other products as discussed above. It also leads to accumulation of substrates Acetyl CoA and Acetoacetyl CoA. These accumulate as long chain CoAs.
KATP channels are octameric proteins present ubiquitously in the human tissue in a stoichiometry of 4:4. They are composed of tetramers in 4:4 ratio with inner core formed by inward rectifier pore of Kir 6.x surrounded by SUR (Sulphonylurea receptor).
Pore forming inward rectifier potassium (Kir) channel is activated by a decrease of ATP/ADP [26] and also by accumulation of LC CoA. Statins exert their observed and well known pleiotropic effects by activating KATP channels.
Studies on the involvement of KATP channels in nociceptive processing are scarce in literature and they have largely been unexplored. KATP channel activation leads to reduction in hyperalgesia. Studies on mice suggest that peripheral nerve activity is modulated by KATP channels in both myelinated and unmyelinated nerve fibres [27].
Activation of KATP channels directly affects VGCC (Voltage Gated Calcium Channels) and leads to their inhibition. Inhibition of these calcium channels should rather alleviate the neuropathic pain as is manifested with the administration of calcium channels inhibitors like gabapentin.
However, a simultaneous deficiency of ubiquinone caused by disturbance in cholesterol biosynthesis is responsible for the neuropathic pain as has been described earlier.
Statins tend to act in a manner which is similar to the action of calcium channel blockers
When the effect of statins was studied on the spontaneous activity of isolated rabbit’s jejunal preparation, it was observed that simvastatin, atorvastatin, and rosuvastatin relaxed the KCl induced and spontaneous contractions. The researchers concluded that stains tend to show effects similar to that of verapamil, a calcium channel blocker [28].
The gastrointestinal symptoms observed in patients on statin therapy may be because of their calcium channel blocking effects [29] Statins and calcium channel blockers have been shown to have synergistic effects [30] in vascular smooth musculature also besides the GI system.
Voltage Gated Calcium Channels (VGCC) are being exploited in chronic pain management. N type VGCC are modulated by morphine, when an inhibition of neurotransmitter release occurs because of G Protein dependent inhibition of calcium currents [31-33] and pain signals are reduced.
Examples of L type VGCC blockers are topiramate, gabapentin and pregabalin which bind to Cav X2 S1 subunit of VGCC [34,35]. Figure 2 illustrates as to how statins inhibit calcium channels indirectly when they activate KATP channels, and therefore their action mimics that of calcium channel inhibitors.
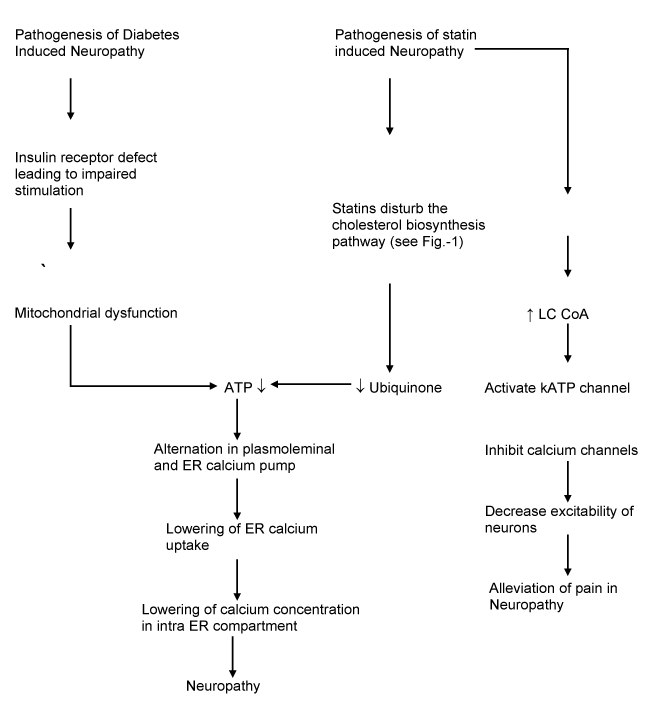
Figure 2: Proposed hypothesis of the neuropathy pathophysiology of statin induced and diabetic neuropathy. ATP depletion is the core reason for this neuropathy.
If a diabetic patient is put on statins, the already depletion ATP is further enhanced leading to more severe neuropathy. This is partially overcome by the calcium channel inhibiting effect of statins.
Expert opinion
Deficiency of ubiquino2021 Copyright OAT. All rights reserveuropathic symptoms in a diabetic patient suffering from neuropathy. Statins use leads to further ubiquinone deficiency in these patients. Statins also activate KATP channels which in turn inhibits calcium channels. This should theoretically be helpful, in a manner similar to what is seen with use of gabapentin, in alleviating neuropathic pain. Diabetic patients already are deficient in Ubiquinone. The ubiquinone becomes further depleted when statins are administered to diabetic patients. The calcium channel inhibitory action of statins, which otherwise would have ameliorated neuropathic pain, is neutralized by ubiquinone deficiency which occurs due to their inhibitory action on cholesterol biosynthesis.
Ubiquinone or coenzyme Q in human mitochondria is called CoQ10 because of the number of isoprene repeats observed in it. CoQ10 levels are highest in organs with a high rate of metabolism like liver, heart and kidney. It acts as a cofactor in the electron transport chain and is involved in the synthesis of ATP and thus is involved in production of energy for cells. It also prevents generation of free radicals. As in evident in Figure-2, a direct deficiency of ATP in diabetic patients may be leading to diabetic neuropathy. However, a further decrease in levels of ATP caused by statin use exacerbates the symptoms of neuropathy.
A reduced CoQ 10 or ubiquinone is not able to act on free radicals to prevent lipid peroxidation and protect mitochondrial DNA. CoQ 10 facilitates electron transfer amongst redox components in the transport chain of electrons. This creates a gradient of protons along the inner membrane of mitochondria. It leads to increased ATP production. Therefore, a decreased production of ATP by interruption of cholesterol biosynthesis by statins occurs. This undesired side effect of statin therapy may be corrected by supplementation of ubiquinone [34-37] Even otherwise, in patients of type 2 diabetes mellitus with neuropathy who are not on statins, ubiquinone / CoQ supplementation should be beneficial in similar manner.
CoQ 10 is available as an ‘over the counter’ product. This has led to an image of CoQ 10 as a ‘non serious’ medication being written for the placebo effect. However, as the above review suggests, CoQ 10 prescription in diabetic neuropathy has a scientific basis. Absence of patent protection for this important molecule has kept away sponsors from conducting large scale trials. Literature is scarce about large scale trials conducted on the effects of CoQ10 administration in patients of neuropathy. Only a few animal studies have been conducted which have indeed demonstrated beneficial effects of CoQ10 [38-42].
References
- Danish National Medicine Division. Laegemiddelstatistik, Denmark, 1994-1998 (in Danish). Copenhagen: Department of Statistics, Medicine Division, 1999.
- Baxter C, Jones R, Corr L (1998) Time trend analysis and variations in prescribing lipid-lowering drugs in general practice. BMJ 317: 1134-1135.
- Scandinavian Simvastatin Survival Group (1994) Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study. Lancet 344: 1383-1389. [Crossref]
- Shepherd J, Cobbe SM, Ford I (1995) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Group. N. Engl J Med 333: 1301-1307. [Crossref]
- Sehra D, Sehra S, Sehra ST (2017) Cardiovascular pleiotropic effects of statins and new onset diabetes: is there a common link: do we need to evaluate the role of KATP channels? Expert Opinion on Drug Safety 16: 823-831. [Crossref]
- Gaist D, Jeppesen U, Andersen M, García Rodríguez LA, Hallas J, et al. (2002) Statins and risk of polyneuropathy, a case control study.. Neurology 58: 1333-1337. [Crossref]
- Jacobs MB (1994) HMG-CoA reductase inhibitor therapy and peripheral neuropathy. Ann Intern Med 120: 970. [Crossref]
- Ahmad S (1995) Lovastatin and peripheral neuropathy. Am Heart J 130: 1321. [Crossref]
- Phan T, McLeod JG, Pollard JD, Peiris O, Rohan A, et al. (1995) Peripheral neuropathy associated with Simvastatin. J Neurol Neurosurg Psychiatry 58: 625-628.
- Ziajka PE, Wehmeier T (1998) Peripheral neuropathy and lipid-lowering therapy. South Med J 91: 776-68.
- Gaist D, García Rodríguez LA, Huerta C, Hallas J, Sindrup SH (2001) Are users of lipid-lowering drugs at increased risk of peripheral neuropathy? Eur J Clin Pharmacol 56: 931-933. [Crossref]
- Otruba P, Kanovsky P, Hlustik P (2011) Treatment with statins and peripheral neuropathy: results of 36- months a prospective clinical and neurophysiological follow-up. Neuro Endocrinol Lett 32: 688-690.
- Emad M, Arjmand H, Farpour HR, Kardeh B (2018) Lipid lowering drugs (statins) and peripheral neuropathy. Electronic Physician 10: 6527-6533.
- McMains PG, Windeback AJ, Kiziltan M (1994) Neuropathy associated with hyperlipidemia. Neurology 44:2185-2186.
- Malik RA, Tesfaye S, Newrick PG, Walker D, Rajbhandari SM, et al. (2005) Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 48: 578-585.
- Sima AA (2004) Diabetic neuropathy in type 1 and type 2 diabetes and the effects of C-peptide. J Neurol Sci 220: 133-136. [Crossref]
- Yagihashi S (1997) Pathogenetic mechanisms of diabetic neuropathy: lessons from animal models. J Peripher Nerv Syst 2: 113-132.
- Polydefkis M, Hauer P, Sheth S, Sirdofsky M, Griffin JW, et al. (2004) The time course of epidermal nerve fibre regeneration: studies in normal controls and in people with diabetes, with and without neuropathy. Brain 127: 1606-1615.
- Thornalley PJ (2002) Glycation in diabetic neuropathy: characteristics, consequences, causes and therapeutic options. Int Rev Neurobiol 50: 37-57.
- Yan SF, Barile GR, D'Agati V, Du Yan S, Ramasamy R, et al. (2007) The biology of RAGE and its ligands: uncovering mechanisms at the heart of diabetes and its complications. Curr Diab Rep 7: 146-153. [Crossref]
- Gover TD, Moreira TH, Weinreich D (2009) Role of calcium in regulating primary sensory neuronal excitability. Handbook of experimental pharmacology 194: 563-587. [Crossref]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, et al. (2005) Silencing of the Cav 3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. The EMBO journal 24: 315-324.
- Wen XJ, Li ZJ, Chen ZX, Fang ZY, Yang CX (2006) Intrathecal administration of Cav3.2 and Cav 3.3 antisense oligonucleotide reverse tactile allogynia and thermal hyperalgesia in rats following chronic compression of dorsal root of ganglion. Acta pharmacologica Sinica 27: 1547-1552. [Crossref]
- Hucho T, Levine JD (2007) Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 55: 365-376. [Crossref]
- Fernyhough P, Calcutt NA (2009) Abnormal calcium homeostatis in peripheral neuropathies. Cell Calcium 47: 130-139.
- Sehra D, Sehra S, Sehra ST (2011) Sulfonylureas: do we need to introspect safety again? Expert Opin Drug Saf 10: 851-861. [Crossref]
- Luu W, Bjork J, Salo E, Entenmann N, Jurgenson T, et al (2019) Modulation of KATP channel activity in the peripheral nervous system reduces mechanical hyperalgesia after nerve injury in mice. Int J Mol Sci 7: 20.
- Ali N, Begum R, Faisal MS, Khan A, Nabi M, et al. (2016) Current statins show calcium channel blocking activity through voltage gated channels. BMC Pharmacology and Toxicology 17: 43. [Crossref]
- Mlodinow SG, Onysko MK, Vandiver JW, Hunter ML, Mahvan TD (2014) Statin adverse effects: sorting out the evidence. J Fam Pract 63: 497-506. [Crossref]
- Clunn GF, Sever PS, Hughes AD (2010) Calcium channel regulation in vascular smooth muscle cells; synergistic effects of statins and calcium channel blockers. Int J Cardiol 139: 2-6.
- Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16: 521-555. [Crossref]
- Schroeder CI, Doering CJ, Zamponi GW (2008) Presynaptic calcium channels: structure, regulators, and blockers. Handb Exp Pharmacol 184: 45-75. [Crossref]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, et al. (1996) Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 380: 258-262. [Crossref]
- Dworkin RH, O’Connor AB, Backonja M (2007) Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 132: 237-251.
- Stahl SM (2004) Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha (2) delta ligands at voltage-gated calcium channels. J Clin Psychiatry 65: 596-597.
- Fernandez-Ayala DJ, Lopebz-Lluch G, Gracia-Valdes M, Arroyo A, Navas P () Specificity of coenzyme Q 10 for a balanced function of respiratory chain and endogenous. Biochim Biophys Acta 1706: 174-83. [Crossref]
- Prakash S, Sunitha J, Hans M (2010) Role of coenzyme Q(10) as an antioxidant and bioenergizer in periodontal diseases. Indian J Pharmacol 42: 334-337. [Crossref]
- Shi TJS, Zhang MD, Zeberg H, Grunler NJ, Liu SuX, et al. (2013) Coenzyme Q10 Prevents peripheral neuropathy and attenuates neuron loss in the db-/ db- mouse, a type 2 diabetes model. PNAS 110: 690-695.
- Sullivan KA (2007) Mouse models of diabetic neuropathy. Neurobiol Dis 28: 276-285.
- Ayaz M, Tuncer S, Okudan N, Gokbel H (2008) Coenzyme Q (10) and alpha lipoic acid supplementation in diabetic rats : Conduction velocity distributions. Methods Find Exp Clin Pharmacol 30: 367-374.
- Robertson DM, Sima AA (1980) Diabetic neuropathy in the mutant mouse [C57BL/ks (db/db)] : A morphometric study. Diabetes 29: 60-67.
- Sima AA, Robertson DM (1978) Peripheral neuropathy in mutant diabetic mouse [C57BL/Ks (db/db)]. Acta Neuropathol 41: 85-89. [Crossref]
- Olivier LM, Krisans SK (2000) Peroxisomal protein targeting and identification of peroxisomal targeting signals in cholesterol biosynthetic enzymes. Biochim Biophys Acta 1529: 89-102. [Crossref]