Epilepsy is a complex neurological disorder affecting 70 million people worldwide. One of the major issues regarding epilepsy management is resistance to treatment. Coupling the mechanisms of drugs action with the individual pathogenetic mechanisms of epilepsy represents the best strategy for personalized and effective medication. To this regard, genome-wide analysis and extensive database can be utilized to characterize the etiopathogenic mechanisms underlying epilepsy in individual patients and identify the most effective drugs.
In the present paper, we present a systems biology approach to interpret the role of Copy Number Variations (CNVs) in a 27-years old male affected by refractory-epilepsy associated with intellectual disability. In addition to identify causative genotype-phenotype correlations, the synergistic integration of genomic data allowed to decipher the chromosomal aberrations that may underly the pharmaco resistant phenotype.
Given the high multifactoriality of drug resistance in epilepsy, the systems biology approach used here could represent a valid tool for its pharmacological management.
Drug-resistant epilepsy, systems biology, intellectual disability, pharmacogenomics, personalized medicine.
CNVs: Copy Number Variations; AED: Anti-Epileptic Drugs; GABA: Gamma Amino-Butyric Acid; SNP: Single Nucleotide Polymorphism; AQ-EQ: Autism Spectrum Quotient- Empathy Quotient; MRI: Magnetic Resonance Imaging; EEG: Electroencephalogram; CT: Computed Tomography; aCGH: Array Comparative Genomic Hybridization; MLPA: Multiplex Ligation-dependent Probe Amplification; NMDA: N-methyl-D-aspartate; CNS: Central Nervous System; ADHD: Attention Deficit Hyperactivity Disorder; OCD: Obsessive-Compulsive Disorder; WRC: WAVE Regulatory Complex; FMRP: Fragile X Mental Retardation Protein; CSWS: Continuous Spike and Wave During Sleep
Epilepsy is the most common neurological disorder affecting almost 70 million people worldwide. The many different forms of epileptic seizures led to classifying epilepsy as a complex and severe disease characterized by different subtypes [1]. This landscape often makes the diagnosis, prognosis and treatment inaccurate and difficult to manage. The resistance to antiepileptic drugs (AED) represents, in particular, the major issue in patient management [2]. About 20 to 30% of epileptic patients, in fact, show uncontrolled, intractable seizures or adverse side effects secondary to treatment. The reasons for this failure are multiple and are attributed to the heterogeneity of the underlying causes, the type and severity of epilepsy, genetic alterations and pharmacokinetics mechanisms.
AEDs are able to suppress epileptic seizures through different mechanistic pathways [3]. The principal pharmacological targets of AEDs are enzymes, transporters, voltage-gated channels, inhibitory neurotransmitters like GABA or excitatory neurotransmitter like glutamate. Despite the differences in their chemical structure, the shared mechanism of action is the inhibitory effect modulated by the binding to specific receptors. The most commonly used AEDs exert a specific action reducing neuronal excitability and decreasing seizures frequency and/or severity [4]. New antiepileptic agents can exert their action indirectly by post-synaptic neuronal inhibition, modifying metabolic pathways or the function of neurotransmitters/receptors that control channel opening and closing [5]. Multiple theories can explain drug resistance, but the gene variants hypothesis remains one of the most accredited since genetic alterations may not only explain the etiopathogenesis of epilepsy, but also affect pharmacokinetics at different levels [6]. Therefore, knowing the pathways and the signaling cascade that regulate the action of antiepileptic drugs can certainly help to identify the reasons for drug resistance.
Nowadays, high-throughput genomic technologies together with bioinformatics approaches allow to define genetic alterations implicated not only in the etiology of epilepsy but also associated to treatment efficacy and adverse drug reactions [7]. A vast number of single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) are known to exert important effects on both pharmacodynamic and pharmacokinetic mechanisms of AEDs as on drug interaction with its target or drug absorption, distribution and excretion from the body [8]. Publicly available databases provide the starting point to investigate chemical and drug's interactions allowing to define biochemical deregulated pathways, gene/protein interactions, chemical/disease and gene/disease relationships, enabling the formulation of hypothesis-driven outcomes [9,10].
The integration of all available information, using a systems biology approach that takes into account the genomic profile of a patient and the whole complexity of refractory epilepsy, may represents an important strategy to identify personalized treatments [11,12]. The principle that is now becoming a "mindset" in research is to consider all parts of a biological system, together with its specific, coordinated and dynamic connections. The integration of chemical, genetic, genomic and clinical data may enable a systems biology approach that offers a comprehensive understanding of epilepsy etio-pathogenesis, elucidating fundamental drug-disease relationships and predicting molecular networks and unsuspected drug interactions [13-15].
Although feasible in research labs, the present challenge is to translate the systems biology approach into clinical practice. The expected benefit is to obtain a patient-specific biological portrait of disease and allow personalized diagnostic, prognostic and therapeutic strategies [16].
In the present paper, we report a case study of a young adult with severe intellectual disability associated with coexisting refractory epilepsy. We provide a proof of concept of how a genomic profile may lead to a double interpretation: the identification of possible etiological genetic variants together with aberrations that can interfere with drug response.
A 27-years old man with intellectual disability and refractory generalized tonic-clonic epilepsy was presented. To patient enrollment, written informed consent to perform a genomic evaluation and for the publication of this case report was obtained. On examination, physical and neurological status was evaluated, and the cognitive phenotype was characterized by a severe intellectual disability and neurobehavioral disorder. The patient exhibited poor social interaction, poor verbal and nonverbal communication and restricted interests. These parameters were evaluated by psychological tests (AQ-EQ, Autism Diagnostic Interview-Revised, Autism Diagnostic Observation Schedule, Raven's Progressive Matrices) [17]. We retrospectively reviewed laboratory and clinical data regarding gender, age, genetic conditions, presence or absence of regression, age of first seizure, treatment with or without response to antiepileptic drugs, magnetic resonance imaging (MRI) and electroencephalographic (EEG) recordings. Seizures started at around age 10 years old; pharmacological treatment, including different types of neuroleptics and antiepileptic drugs (Aripiprazole, Ziprasidone, Delorazepam, Lormetazepam, Quetiapine, Flurazepam, Promazine, Triazolam, Levetiracetam, Clonazepam, and Lamotrigine) in mono- or polytherapy, was reported ineffective. Sedative drugs were able to decrease seizures but did not stop it. EEG pattern showed continuous spikes and waves discharges at posterior head regions. MRI and CT of the brain revealed no significant variations. Laboratory data revealed high levels of vanillylmandelic acid, low level of DHEA-S and normal levels of serum cortisol and testosterone. At the age of 16, an 4x44k array-based comparative genomic hybridization (aCGH) analysis was performed on patient’s DNA. The report pointed out a deletion of about 1.5 Mb region on chromosome 15q11.2 [19.109.124-20.636.537; arr(h18)] without listing any gene included in this deletion or associating it to any phenotype. A multiplex ligation-dependent probe amplification (MLPA) assay did not detect deletions and/or duplications in subtelomeric regions of genomic DNA.
To identify possible genetic etiological variants together with aberrations that can interfere with drug response, we performed aCGH analysis using an 8x60k Whole Genome SurePrint G3 microarray (Agilent Technologies, Santa Clara, CA). High molecular weight DNA was extracted from blood using an automatic extractor (Qiagen, Hilden, Germany), quantified by ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and assessed for quality by microcapillary electrophoresis on a 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). The aCGH analysis was performed according to the manufacturer’s guidelines provided by Agilent Technologies. Features were extracted with the Feature Extraction v5.0.0.1 software program, using default settings. To screen copy number alterations in the genome, we used the Aberration Detection Method 2 (ADM-2) algorithm developed by Agilent and included in the Agilent Genomic Workbench (Agilent Technologies, Santa Clara, CA, USA) for CGH analysis. Data were transformed as log2 ratios (Cy3 intensity/Cy5 intensity). The minimum number of 3 consecutive probes with the same copy number change and a minimum absolute log2 ratio for region >0.5 were chosen to define an aberration region. The threshold of 6 was set, as recommended by the array manufacturer.
Following the general workflow of Systems Biology, clinical findings, experimental data reported in scientific literature and publicly available database (DrugBank, DGIdb) were interrogated to study drug actions, molecular targets of chemical compounds, associations between genes and drugs. MetaCore software (Clarivate Analytics, Philadelphia, United States) was used to perform a functional enrichment analysis and to investigate molecular interactions and networks in our lists of genes. Moreover, to interpreter the variants with respect to the biological processes, a systematic review of articles was performed.
To evaluate the genetic etiological variants together with aberrations that can interfere with drug response, we applied a systems biology approach based on aCGH analysis in a 27-years old man with intellectual disability and drug resistant epilepsy.
Genome-wide characterization of copy number variations (CNVs) highlighted several microdeletions and microduplications (Table 1). Among them, the CNV at the Chr15q11.1-11.2 is linked to neurodevelopmental and motor delays, psychiatric problems, attention deficit and autism spectrum disorders [18-21].
Table 1. Pathogenic Copy Number Variants in a patient with refractory epilepsy and intellectual disability
Chr |
Start-Stop |
Cytoband |
Size (Kb) |
Amp/Del |
Annotations |
Chr6 |
37979601-38068930 |
p21.2 |
89.33 |
0.917 |
ZFAND3 |
Chr8 |
10029423-10126317 |
p23.1 |
96.895 |
-0.895 |
MSRA |
Chr9 |
29136982-31263003 |
P21.1 |
2126.022 |
0.751 |
LINGO2, LINCO1242 |
Chr15 |
22784523-23179948 |
q11.2 |
395.426 |
-0.814 |
NIPA1, NIPA2, CYFIP1, TUBGCP5 |
Together with the aberration on Chr15, our analysis revealed the involvement of other genes: ZFAND3, MSRA, LINGO2 and FAM30A on Chr6, Chr8, Chr9 and Chr14, respectively.
To translate the role of gene aberrations in the context of biological phenomena and, in particular, to put them in the context of the disease, we analyzed the whole genome profile obtained by aCGH using MetaCore. The aCGH data were merged with information related to pharmacokinetics of AED considering the multiple protein effectors and biochemical pathways involved. The explorative study performed with an interacting network approach, allowed to better investigate the implications of genetic aberrations in the etio-pathogenesis of epilepsy and the potential connections with the response to specific treatments (Figure 1).
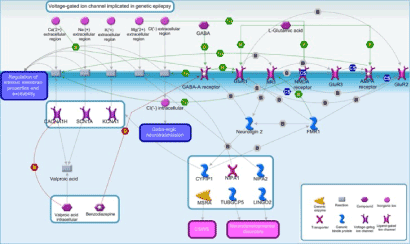
Figure 1. Possible mechanisms underlying refractory epilepsy and intellectual disability. The proposed scheme is a general summary of CNVs detected by aCGH. TUBGCP5, CFYIP1, NIPA1, and NIPA2 genes are located in the chromosome 15q11.2 region and are known to cause neurological, cognitive or behavioral deficits. Chromosome abnormalities identified may contribute to excitatory/inhibitory status imbalance and resistance to antiepileptic drugs
Among the different chromosomal aberrations detected in this patient, we will first focus on a deletion in the long arm of chromosome 15. This region, encompassing a 400-kb, contains low copy repeats known as breakpoint 1 and 2 (bp1 and bp2) [22] and includes four genes: non imprinted in Prader-Willi/Angelman syndrome 1 and 2 (NIPA1 and NIPA2), tubulin gamma complex associated protein 5 (TUBGCP5), and cytoplasmic FMR1 interacting protein 1 (CYFIP1), which is highly expressed in central nervous system neuronal tissues [23] and unmistakably involved in development of neurological and psychiatric disorders [24-26] (Figure 2).
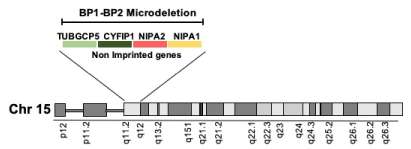
Figure 2. The 15q11.2 BP1-BP2 microdeletion. The area between BP1 and BP2 encompasses a 400 Kb region and includes the TUBGCP5, CFYIP1, NIPA1, and NIPA2 genes
NIPA1 and NIPA2 are integral membrane proteins responsible for Mg2+ transport. Unlike other members of the NIPA family, NIPA2 is a highly selective Mg2+ transporter [27] with a crucial role in Mg2+ metabolism and regulation of intracellular environment conservation. Mg2+ exerts a voltage-dependent block of NMDA receptors activity, regulating the opening of Ca2+ channel and, therefore, neuronal excitability and development of epileptic seizures [28]. Many studies reported that Mg2+ excess causes functional disturbances of Central Nervous System (CNS), while its deficiency produces increased neuronal activity with convulsions [29,30]. Therefore, alterations in NIPA2, resulting in decreased Mg2+ intracellular concentration, may be correlated to NMDA receptor hyperactivity and seizures development [31]. Xie, et al. reported an association between three NIPA2 mutations (p.I178F, p.N244S and p.N334_E335insD), which decrease protein expression in plasma membrane, and childhood absence epilepsy. These alterations entailed a reduction in Mg2+ intracellular concentration, causing increased NMDA receptor activity and neuronal hyper-excitation [31].
TUBGCP5 contributes to the formation of the gamma tubulin complex involved in the microtubule nucleation at the centrosome. This gene is highly expressed in brain, chiefly in the subthalamic nuclei, and its alterations were linked to attention deficit hyperactivity disorder (ADHD) and obsessive-compulsive disorder (OCD) [32].
CYFIP1 is a component of the WAVE Regulatory Complex (WRC) that includes also CYFIP2, WAVE, NAP1, ABI1, HSPC300. It is involved in the activation of WRC and cytoskeletal remodelling [33]. A correlation between the reduction of CYFIP1 and the expression of other WRC members has been reported [34]. Alterations in CYFIP1 expression affect the transcriptional control of cytoskeletal genes, influencing neuronal morphology, number of dendritic spines and remodelling of the synaptic connections, molecular functions that are all deregulated in neurodegenerative diseases [35]. CYFIP1 interacts with the fragile X mental retardation protein (FMRP) encoded by FMR1, whose mutations cause the Fragile X syndrome, characterized by attention deficit, autistic behavior and epileptic seizures [36]. Experimental evidences suggested involvement of CYFIP1 in maintaining the balance between the excitatory/inhibitory state of the CNS [37]. Different in vivo studies reported that a reduction in the expression of the α1, α3, α4, α5, β1, β2, γ1, γ2, and δ subunits in fragile X mouse models, impair the inhibitory action of GABAergic signalling [38-40]. CYFIP1 knockout models show elevated expression of post-synaptic GABAA receptor β-subunits with the subsequent increase in synaptic inhibition. In contrast, enhanced levels of CYFIP1 were correlated to a loss of GABAA receptor γ2-subunit on the postsynaptic surface [37]. Although deregulated expression of CYFIP1 is not sufficient to explain the resistance to drug treatment in epileptic subjects, it was observed that its knockdown alters disease-gene networks related to the 15q11.2 BP1-2 deletion. Finally, regulation of epilepsy associated genes, such as CACNA1H, KCNA1, and SCN1A, is influenced by CYFIP1 [26].
Analysis of genomic data led to the hypothesis that the possible deficit of Mg2+ caused by the loss of NIPA2, the conceivable reduction of GABAA receptor subunit expression and the CACNA1H, KCNA1, and SCN1A deregulation associated with the CYFIP1 deletion, may contribute to the excitatory/inhibitory status imbalance as well as the altered response to antiepileptic drugs. In particular, increased NMDA synaptic and extra-synaptic currents, linked to aberrations on Chr15q11.1-11.2, may impact on GABAA receptors trafficking (41), leading to internalization of synaptic GABAA receptor [42]. NMDA activation and the permanent alterations in Ca2+ homeostatic mechanisms may result in chronically elevated basal Ca2+ and alteration of the post-synaptic Ca2+ balance triggering post-synaptic signaling. This may end up to the failure of inhibition and the emergence of self-sustaining seizures.
Our study revealed additional imbalances on other chromosomal segmentations that may also contribute to the atypical phenotype. Indeed, we detected CNVs in genes previously implicated in other neurodevelopmental disorders and epilepsy, such as MSRA, ZFAND3, LINGO2 and FAM30A. In particular MSRA, located on Chr 8p23.1, was associated to Epileptic Encephalopathy With Continuous Spike and Wave During Sleep (CSWS), a very rare form of epilepsy refractory to conventional anti-epileptic drug therapy [43]. ZFAND3 on Chr 6p21.2 and LINGO2 on Chr 9p21.1 were linked to autism and neurodevelopmental disorders, respectively [44,45].
Overall, the results obtained show how genome-wide data may be helpful to identify biological pathways and dysregulated interactions underlying complex diseases. Together with the identification of etiological genetic variants, the results show how the analysis of a genomic profile may also help to elucidate the mechanism of refractory epilepsy.
The Systems biology approach here proposed, may help to elucidate the molecular causes of complex disorders, such as epilepsy and neurodevelopmental disorders, and guide to drug therapy [46]. Over-imposing the genomic portrait of a patient to drug mechanism of action, active chemical ligands and biological targets is nowadays feasible and may have important implications if translated into clinical care of refractory epilepsy. Personalized medicine of refractory epilepsy is just a few steps forward and requires physicians to apply a holistic approach, where diagnosis of etio-pathogenetic mechanisms and therapy are integrated.
Ethics approval and consent to participate
This study was carried out in accordance with the recommendations of Ethics committee Catania 1, University Hospital Policlinico Vittorio Emanuele, with written informed consent from all subjects. The patient gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethics committee Catania 1. Authorization n.39/2018/CA.
Consent
Informed consent was obtained from the patient's parent for publication of this report and any accompanying images.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the CNR Research Projects No. DSB.A009.001.018 and DSB.AD008.456.
MG carried out experimental work, analyzed the data and wrote the manuscript; GN participated in data analysis and in writing the manuscript; FLC participated in revising the manuscripts; SC conceived, directed and supervised the project.
Authors gratefully acknowledge the IRIB-CNR staff and Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo, Francesco Marino for their administrative and technical support.
- Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, et al. (2014) ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55: 475-482. [Crossref]
- Kalilani L, Sun X, Pelgrims B, Noack-Rink M, Villanueva V (2018) The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 59: 2179-2193. [Crossref]
- Dalal KS, Bridgeman MB (2016) Antiepileptic drugs. Nursing 46: 70. [Crossref]
- White HS, Smith MD, Wilcox KS (2007) Mechanisms of action of antiepileptic drugs. Int Rev Neurobiol 81: 85-110. [Crossref]
- Rogawski MA, Loscher W, Rho JM (2016) Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb Perspect Med. 6: 5. [Crossref]
- Ieiri I, Takane H, Hirota T, Otsubo K, Higuchi S (2006) Genetic polymorphisms of drug transporters: pharmacokinetic and pharmacodynamic consequences in pharmacotherapy. Expert Opin Drug Metab Toxicol. 2: 651-674. [Crossref]
- Mirza N, Vasieva O, Marson AG, Pirmohamed M (2011) Exploring the genomic basis of pharmacoresistance in epilepsy: an integrative analysis of large-scale gene expression profiling studies on brain tissue from epilepsy surgery. Hum Mol Genet 2011 Nov 15;20(22):4381-94. [Crossref]
- Tate SK, Sisodiya SM (2007) Multidrug resistance in epilepsy: a pharmacogenomic update. Expert Opin Pharmacother 8: 1441-1449. [Crossref]
- Caroli J, Sorrentino G, Forcato M, Del Sal G, Bicciato S (2018) GDA, a web-based tool for Genomics and Drugs integrated analysis. Nucleic Acids Res 46: W148-W156. [Crossref]
- Chua HN, Roth FP (2011) Discovering the targets of drugs via computational systems biology. J Biol Chem. 286: 23653-23658. [Crossref]
- Civelek M, Lusis AJ (2014) Systems genetics approaches to understand complex traits. Nat Rev Genet 15: 34-48. [Crossref]
- Villoslada P, Steinman L, Baranzini SE (2009) Systems biology and its application to the understanding of neurological diseases. Ann Neurol 65: 124-139. [Crossref]
- Harrill AH, Rusyn I (2008) Systems biology and functional genomics approaches for the identification of cellular responses to drug toxicity. Expert Opin Drug Metab Toxicol. 4: 1379-1389. [Crossref]
- Bunnik EM, Le Roch KG (2013) An Introduction to Functional Genomics and Systems Biology. Adv Wound Care 2: 490-498. [Crossref]
- Myers CT, Mefford HC (2015) Advancing epilepsy genetics in the genomic era. Genome Med 7: 91. [Crossref]
- Leyens L, Richer E, Melien O, Ballensiefen W, Brand A (2015) Available Tools to Facilitate Early Patient Access to Medicines in the EU and the USA: Analysis of Conditional Approvals and the Implications for Personalized Medicine. Public Health Genomics 18: 249-259. [Crossref]
- de Bildt A, Sytema S, Zander E, Bolte S, Sturm H, et al. (2015) Autism Diagnostic Interview-Revised (ADI-R) Algorithms for Toddlers and Young Preschoolers: Application in a Non-US Sample of 1,104 Children. J Autism Dev Disord 45: 2076-2091. [Crossref]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, et al. (2009) 15q13. 3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet 41: 160. [Crossref]
- Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, et al. (2009) Familial and sporadic 15q13. 3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet 18: 3626-3631. [Crossref]
- De Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, et al. (2009) Recurrent microdeletions at 15q11. 2 and 16p13. 11 predispose to idiopathic generalized epilepsies. Brain. 133: 23-32. [Crossref]
- Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, et al. (2010) Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet 6: e1000962. [Crossref]
- Butler MG (2017) Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. Journal of intellectual disability research. J Intellect Disabil Res 61: 568-579. [Crossref]
- Chai J, Locke D, Greally J, Knoll J, Ohta T, et al. (2003) Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet 73: 898-925. [Crossref]
- Picinelli C, Lintas C, Piras IS, Gabriele S, Sacco R, et al. (2016) Recurrent 15q11. 2 BP1‐BP2 microdeletions and microduplications in the etiology of neurodevelopmental disorders. Am J Med Genet B Neuropsychiatr Genet 171: 1088-1098. [Crossref]
- Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, et al. (2015) 15q11. 2 microdeletion (BP1–BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet 58: 140-147. [Crossref]
- Nebel RA, Zhao D, Pedrosa E, Kirschen J, Lachman HM, et al. (2016) Reduced CYFIP1 in human neural progenitors results in dysregulation of schizophrenia and epilepsy gene networks. PloS One 11: e0148039. [Crossref]
- Goytain A, Hines RM, Quamme GA. Functional characterization of NIPA2, a selective Mg2+ transporter. Am J Physiol Cell Physiol 295: C944-C53. [Crossref]
- Cao H-Y, Jiang Y-W, Liu Z-W, Wu X-R (2003) Effect of recurrent epileptiform discharges induced by magnesium-free treatment on developing cortical neurons in vitro. Dev Brain Res 142: 1-6. [Crossref]
- Riggs JE (2002) Neurologic manifestations of electrolyte disturbances. Neurol Clin 20: 227-239. [Crossref]
- Castilla‐Guerra L, Fernández‐Moreno MdC, López‐Chozas JM, Fernández‐Bolaños R (2006) Electrolytes disturbances and seizures. Epilepsia 47: 1990-1998. [Crossref]
- Xie H, Zhang Y, Zhang P, Wang J, Wu Y, et al. (2014) Functional study of NIPA2 mutations identified from the patients with childhood absence epilepsy. PloS one 9: e109749. [Crossref]
- De Wolf V, Brison N, Devriendt K, Peeters H (2013) Genetic counseling for susceptibility loci and neurodevelopmental disorders: the del15q11. 2 as an example. Am J Med Genet A 161: 2846-2854. [Crossref]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, et al. (2010) Structure and control of the actin regulatory WAVE complex. Nature 468: 533. [Crossref]
- Abekhoukh S, Sahin HB, Grossi M, Zongaro S, Maurin T, et al. (2017) New insights into the regulatory function of CYFIP1 in the context of WAVE-and FMRP-containing complexes. Dis Model Mech 10: 463-474. [Crossref]
- Pathania M, Davenport E, Muir J, Sheehan D, López-Doménech G, et al. (2014) The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl Psychiatry 4: e374. [Crossref]
- Schenck A, Bardoni B, Moro A, Bagni C, Mandel J-L (2001) A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A 98: 8844-8849. [Crossref]
- Davenport EC, Szulc B, Drew J, Taylor J, Morgan T, et al. (2018) Correct CYFIP1 dosage is essential for synaptic inhibition and the excitatory/inhibitory balance. bioRxiv 303446. [Crossref]
- d'Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, et al. (2006) Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res 1121: 238-245. [Crossref]
- Curia G, Papouin T, Séguéla P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex 19: 1515-1520. [Crossref]
- Sabanov V, Braat S, D'andrea L, Willemsen R, Zeidler S, et al. (2017) Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice. Neuropharmacology 116: 71-81. [Crossref]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 25: 7724-7733. [Crossref]
- Wasterlain CG, Naylor DE, Liu H, Niquet J, Baldwin R. Trafficking of NMDA receptors during status epilepticus: therapeutic implications. Epilepsia 6:78-80. [Crossref]
- Yuan Q, Li F, Zhong H. Early diagnosis, treatment and prognosis of epilepsy with continuous spikes and waves during slow sleep. Int J Clin Exp Med 8: 4052. [Crossref]
- Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, et al. (2012) A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 2: 1665-1685. [Crossref]
- Matsunami N, Hadley D, Hensel CH, Christensen GB, Kim C, et al. (2013) Identification of rare recurrent copy number variants in high-risk autism families and their prevalence in a large ASD population. PloS one 8: e52239. [Crossref]
- Gonzalez-Angulo AM, Hennessy BT, Mills GB (2010) Future of personalized medicine in oncology: a systems biology approach. J Clin Oncol 28: 2777-2783. [Crossref]