Abstract
The ETS transcription factors regulate expression of genes involved in normal cell development, proliferation, differentiation, angiogenesis, and apoptosis, consisting of 28 family members in humans. Dysregulation of these transcription factors facilitates cell proliferation in cancers, and several members participate in invasion and metastasis by activating gene transcription. ETS1 and ETS2 are the founding members of the ETS family and regulate transcription by binding to ETS sequences. They are both involved in oncogenesis and tumor suppression depending on the biological situations used. The essential roles of ETS proteins in human telomere maintenance have been suggested, which has been linked to creation of new Ets binding sites. Recently, preferential binding of ETS2 to gain-of-function mutant p53 and ETS1 to wild type p53 (WTp53) have been suggested, raising the tumor promoting role for the former and tumor suppressive role for the latter. The oncogenic and tumor suppressive functions of ETS1 and 2 proteins have been discussed.
Key words
ETS1, ETS2, telomerase, mutant p53, expression, cancer
Introduction
The oncogene v-ets was originally discovered as a component of a chimeric gene along with a truncated v-myb gene, present in the genome of E26, an avian leukosis virus (Figure 1A) [1-3]. Members of the ETS gene family have been cloned and sequenced from a variety of species ranging from human to Drosophila. Ets proteins are highly conserved through different species [4].
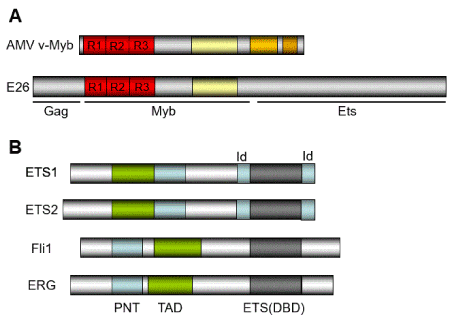
Figure 1. The structure of ETS proteins. A) The structure of AMV v-Myb and E26 retrovirus. R1, R2, and R3 are Myb-like repeats. The E26 virus consists with Gag, Myb, and Ets-like regions (1-3, 52-54). B) The structure of ETS proteins reviewed in this article. The domain structures for ETS1, ETS2, Fli1, and ERG are shown. The ETS domain that is essential for DNA-binding is shown in dark box. The DNA-binding by the ternary complex factor (TCF) subfamily of ETS-domain transcription factors (4) is tightly regulated by intramolecular and intermolecular interactions. The helix-loop-helix (HLH) -containing Id proteins (138) are trans-acting negative regulators of DNA binding by the TCFs. Inhibition is mediated by direct interactions of the Ids with the ETS DNA-binding domain of the TCFs. PNT: pointed domain, TAD: transactivation domain, ID: Id-interaction domain.
The ETS family of transcription factors have the conserved primary sequence of their DNA-binding domains (Figure 1; ETS1/2, ERG, and FLI1 are shown). The human ETS factors are classified into 11 subgroups based upon ETS domain sequence homology: ETS1/2, ERG, PEA3 (ETV1/4/5), TCF (ELK1/3/4), GABP (GABPA: GA [purine] -binding protein alpha chain), ELF1/2/4, SPI1 (SPI1/B/C), TEL (ETV6/7), ERF (ERF, ETV3, ETV3L), FLI1 and FEV [4]. In addition, a subset of four ETS family genes (ELF3, ELF5, EHF, SPDEF) has been characterized as placenta-specific subgroup based upon their restricted expression to tissues with high epithelial cell content [4-6], totally 28 ETS proteins in humans.
The ETS transcriptions factors regulate numerous genes by binding to winged helix-turn-helix GGAA/T core of DNA-binding, and are involved in cell proliferation, differentiation, development, stem cell development, transformation, angiogenesis, and apoptosis [7-13]. ETS1 and ETS2 are representative members of the ETS family of transcription factors and are downstream effectors of the RAS/RAF/ERK pathway [14-19] (Figure 2). The pointed domain is at the N-terminal end (Figure 1, Pnt-P2 and Yan domain [PNT] in [20]). Overexpression of dominant-negative forms of several ETS factors, including ETS1 or ETS2, block Ras transformation [16,21,22] suggesting that ETS family members play a crucial role in this process. However, specific deletions of ETS family members are more accurate approaches for the understanding of functions of ETS members in Ras transformation [23]. It has been reported that aberrant activation of the ETS family transcription factors play roles throughout all stages of tumorigenesis [24]. Specifically in solid tumors, gene rearrangement/amplification, feed-forward growth factor signaling loop, formation of gain-of-function co-regulatory complexes, and novel cis-acting mutations in ETS target gene promoters can result in increased ETS activity [24]. The ETS signaling enhances tumorigenesis through different mechanisms such as lineage specification, increased self-renewal, and genomic instability confirming the proto-oncogenic roles of ETS proteins in cancer.
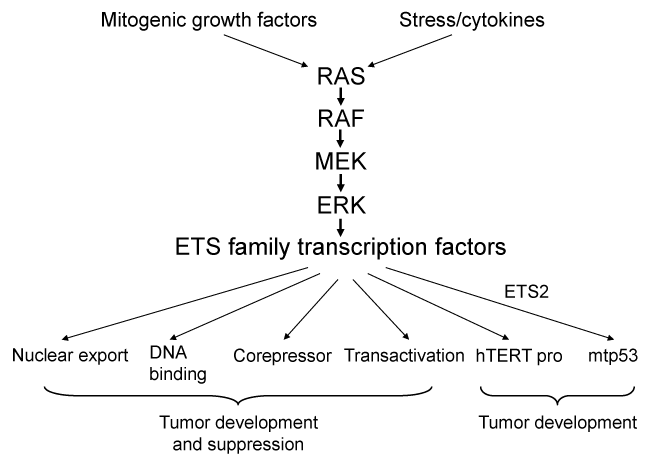
Figure 2. Oncogenic and tumor suppressive activities of ETS proteins. Both mitogenic signals stemming from growth factors and stress signals stimulate the RAS-RAF-MEK-ERK pathway to activate the ETS transcription factors. ETS proteins show oncogenic or tumor suppressive activities by increased nuclear transport [178,179], enhanced DNA binding of nuclear proteins [180,181], recruitment of co-repressors [182,183], and gene transactivation [184,185] involved in cell proliferation, apoptosis, angiogenesis. Mutation of hTERT promoter created novel ETS binding sites, which contributes increased telomerase activity in cancer cells. Mutant p53 which is overexpressed in half of human cancer cells, bind to ETS2 to stabilize the protein and reveal its oncogenic activity.
It was reported that that the ETS2 gene was located about 17 cM from the breakpoint of common t(8;21) translocation found in acute myelogenous leukemia (AML) [25,26], esp. M2 subclass. Then the chimeric gene at t(8;21) translocation was AML1-ETO was cloned and characterized [27,28]. Although the chimeric gene responsible for acute myeloid leukemia was did not contain ETS, the ETS family ERG1 was situated just proximal to ETS2 at 21q22.3 [29,30] (Figure 1B). They found that the ERG1 gene was translocated from chromosome 21 to chromosome 8 in the t(8;21)(q22;q22) [30]. The ERG protein was considerably more stable than the short-lived ETS1 and ETS2 proteins with a half-life of 21 hours. It was then reported that ERG2 is a nuclear phosphoprotein bound to purine-rich sequences [31]. The ERG2 protein was a sequence-specific, DNA-binding protein and is expressed at higher levels in early myeloid cells than in mature lymphoid cells, acting as a regulator of genes required for maintenance and differentiation of early hematopoietic cells [31].
Human telomerase reverse transcriptase gene (TERT) encodes a rate-limiting catalytic subunit of telomerase essential for genomic integrity [11,32-34]. TERT expression is repressed in somatic cells except for self-renewing proliferative cells and tumor cells [32]. Thus, immortality associated with cancer cells has been thought to be caused by telomerase overexpression. Importantly, the newly described germline and recurrent somatic mutations in melanoma [35] and other cancers [36] in the TERT promoter that create de novo ETS/TCF binding sites, provide an insight into the possible cause of tumor-specific increased TERT expression [32,33,37].
Most mutant p53 (mtp53) expressed in cancer cells has gain-of-function (GOF) associated with genomic instability, increased tumorigenicity and stem cell-ness, invasiveness/metastasis, and resistance to chemo/radiation therapy [38]. In cancer cells expressing GOF mutant p53, groups of genes that belong to the CREB1 [39], NRF2 [40], c-Myc [41,42], Ets1/2 [43,44], networks as well as those that are involved in chromatin methylation/acetylation (MLL1/2, MOZ; 45) are transactivated (mtp53 reviewed in [38,44,46]. Preferential binding of ETS1 to WTp53 and ETS2 to mtp53 have been reported, suggesting tumor-suppressive function for the former and oncogenic function for the latter, which is discussed in this review.
The ETS gene often fuses with others as a result of translocation. For instance, ETS1 is found in breakpoints such as t(9;11)(p22;q23) and t(4;11)(q21;q23) that are often found in human leukemias [47,48]. It is often overexpressed in human cancer with Cyclin D1 [49], p16INK4a [50] and telomerase [51] overexpression, and thus is heavily involved in human carcinogenesis). Human chromosomal translocations involving ETS family genes have been amply reported on ERG:FUS (acute leukemia), EWS:FLI1 (Ewing sarcoma), ERG:TMPRSS2 (prostate cancer), which will be reviewed in a different paper since Ets is such a big family. Here we will focus on Ets1 and Ets2 in carcinogenesis.
Ets1 and Ets2
Gene cloning and the protein structure
Watson et al. cloned and sequenced ETS1 and ETS2 cDNA clones obtained from human and mouse [52] (Figure 1B). In mammals, the Ets sequences are located on two separate chromosomal loci, called Ets1 and Ets2. To determine the structure and open reading frames of these two genes, they sequenced human Ets1 and Ets2 cDNA clones obtained from both human and mouse. The human ETS1 gene encodes a protein of 441 amino acids. This protein is greater than 95% identical to the chicken c-Ets1 gene product. Thus, the human ETS1 gene is homologous to the chicken c-Ets1 gene, the proto-oncogene that the E26 virus. Human and mouse Ets2 cDNA clones are closely related to Ets1 having open reading frames for proteins of 469 and 468 residues, respectively. By in situ hybridization, de Taisne, et al. [53] mapped human oncogene ETS1 to chromosome 11q23-q24. Next year, Watson et al. identified 2 distinct genomic DNAs homologous to the Ets region of the transforming gene of avian erythroblastosis virus, E26 [54] (Figure 1A).
The ETS1 and ETS2 genes are located on the same chromosome and are coordinately transcribed in avians [54]. Whereas the chicken Ets protein distributes equally between the cytoplasm and nucleus, the human ETS1 protein is cytoplasmic while the ETS2 protein is nuclear suggesting that they have different biologic functions [55]. ETS1 is a 441-amino acid protein that contains an N-terminal domain with PNT implicated in the self-association of oncoproteins and a C-terminal DNA-binding domain (Figure 1B). Of note, it has a MAPK phosphorylation site at Thr38 that mediates transcriptional regulation [56]. Splice variants of ETS1 has been characterized: the full-length ETS1 encodes a 51-kD protein (ETS1-p51) while ETS1DVII lacks exon 7 and encodes ETS1-p42 [57]. The latter lacks the N-terminal inhibitory domain found in ETS1-p51, but has unique DNA-binding and transcriptional properties and regulates different target genes [57]. Laitem, et al. identified a novel variant of Ets1D(III–VI), resulting from the alternative splicing of exons III to VI encoding a 27 kDa isoform [57]. Ets1-p27 lacks the Thr38 residue, PNT, and the transactivation domain, all of which are required for the transactivation by Ets1. They also showed that Ets1-p27 binds DNA in the same way as Ets1-p51 does and induces the translocation of Ets1-p51 from the nucleus to the cytoplasm thereby constituting a dominant-negative of Ets1-p51 [57].
Watson, et al. [54] mapped the ETS2 gene to chromosome 21q22.1-q22.3 by in situ hybridization. Mavrothalassitis [58] reported that the ETS2 gene has no TATA box or CAAT box in its promoter. Then Owczarek et al. determined that the ETS2 gene spans 17.6 kb and has a major CpG island at its 5’ untranslated region [59]. The product, ETS2 protein consists of 469 amino acids with an N-terminal pointed domain and a C-terminal DNA-binding domain [60] (Figure 1B). It also has a MAPK phosphorylation site at Thr72 that likely mediates transcriptional regulation. In short, the structure for ETS1 and ETS2 is very similar (Figure 1B).
Mice model for Ets1-deficiency
T cells go through a number of stages before final differentiation into single-positive CD4 (+) or CD8 (+) α (TCRα) T lymphocytes. The pre-T-cell receptor (TCR) stages include four CD4(-)/CD8(-): double-negative (DN) stages. Namely, DN1 (CD44(+)/CD25(-)), DN2 (CD44(+)/CD25(+)), DN3 (CD44(-)/CD25(+)), and DN4 (CD44(-)/CD25(-)) before differentiation to the double-positive (DP) (CD4(+)/CD8(+)) stage. Eyquem, et al. [61] created mice deficient for the Ets1 transcription factor to determine its role in transition from DN3 to DN4, inhibition of DN cell apoptosis, cellular expansion, and allelic exclusion at the TCRβ locus. Although Ets1-/- embryos were present up to day 18.5 postcoitus in a Mendelian ratio, only 2% of mice were Ets1-/- by 3 weeks of age suggesting most of Ets1-deficiency was embryonic lethal [61]. They found that inactivation of Ets1 impaired the development of DN3 into mature thymocytes and induced an elevated rate of cell death in the DN4 subset [61]. This defect was specific to the α/β lineage because γ/δ T cells maturated efficiently. The percentage of thymocytes co-expressing two different TCRβ chains was increased in the Ets1-deficient background. These data identified Ets1 as a critical transcription factor for pre-TCR functioning and for allelic exclusion at the TCRβ locus [61].
In 2005, Zhan, et al. reported significantly reduced arterial wall thickening, perivascular fibrosis, and cardiac hypertrophy Ets1-deficient mice in response to angiotensin II [62]. The induction of two known targets of ETS1, CDKN1a (p21CIP1) and Plasminogen Activator 1, by angiotensin II was markedly decreased in the aorta of Ets1-null mice compared with wild types [62]. The expression of Monocyte Chemotactic Protein 1 was also reduced, resulting in significantly diminished recruitment of T cells and macrophages to vessel walls. They concluded that ETS1 has an essential role as a transcriptional mediator of vascular inflammation and remodeling in response to angiotensin II [62].
Recent researches have demonstrated positive roles of aberrant splicing in carcinogenesis [63-71]. The full-length Ets1 (p51-Ets1) and Ets1ΔVII (p42-Ets1) isoforms [57] have key distinctions regarding protein-protein interactions, DNA-binding kinetics, and transcriptional target specificity [72]. Splice variant-specific gene targeting studies showed that the Ets1ΔVII mutants demonstrate lymphocyte maturation defects associated with aberrant regulation of p16Ink4a, p27Kip1, and CD44. Thus, a balance in the differential regulation of Ets1 isoforms represents a potential mechanism in the control of lymphoid maturation and homeostasis [72,73].
Mice model for Ets2 overexpression and deficiency
Expression of ETS2 is observed in a variety of cell types. During murine development, it is highly expressed in newly forming cartilage including skull precursor cells, and vertebral primordia [74]. Sumarsono, et al. created transgenic mice to investigate the consequences of overexpression of Ets2 [74]. The mice with less than 2-fold Ets2 overexpression in particular organs developed neurocranial, visceral cranial, and cervical skeletal abnormalities [74]. Of note, these abnormalities had similarities with those found in trisomy 16 humans with Down’s syndrome, in which the gene dosage of ETS2 was increased. Thus, ETS2 has a role in skeletal development and that overexpression is contributed skeletal abnormalities found in Down’s syndrome [75]. This is a nice study that link ETS2 with human disease.
They also reported that overexpression of ETS2 resulted in apoptosis [75]. Transgenic mice overexpressing ETS2 developed a smaller thymus and lymphocyte abnormalities, similar to features observed in Down’s syndrome. Importantly, increased apoptosis correlated with increased expression of p53, and alterations in downstream targets in the p53 pathway [75] suggesting that ETS2 stimulates p53. In HeLa cells, transfection with wild type (WT) p53 enabled ETS2 overexpression to induce apoptosis suggesting crosstalks between ETS2 and p53. Furthermore, crossing the ETS2-transgenic mice with p53-/- mice rescued the thymic apoptosis phenotype [75]. These are extremely important findings in vivo since Ets2 binds to mtp53 and is an essential transcription factor for mtp53 to revel its oncogenic activity (explained later in detail). In conclusion, overexpression of ETS2 induces apoptosis dependent on p53.
Inactivation of Ets2 in mice by deletion of the Ets domain (Ets2db1) caused embryonic lethality before E8.5 [76]. Analysis of the mutant Ets2db1/db1 conceptuses (products of conception) revealed deficiencies in trophoblastic tissues, including a smaller ectoplacental cone and absence of chorion formation. Further analysis has revealed that many Ets2db1/db1 conceptuses fail to form extraembryonic ectoderm [77]. Then Wen, et al. [78] created an Ets2 conditional knockout allele (Ets2flox) in mice. An epiblast-specific Cre line, Mox2-Cre, was crossed with Ets2flox/wt mice to delete Ets2 in germ and embryonic cells [79]. However, the resulting recombined allele designated Ets2db2, caused recessive embryonic lethality around E8 when bred to homozygosity. Ets2db2/db2 embryos at E7.5 were very similar to the Ets2db1/db1 mutants [76] with much smaller conceptuses and unusual cone-shaped yolk sacs. They showed that inactivation of Ets2 resulted in trophoblast stem (TS) cell slower growth, increased expression of differentiation-associated genes, and decreased expression of genes implicated in TS self-renewal [77]. In conclusion, Ets2 contributes to the regulation of genes important for maintaining the undifferentiated state of TS cells and is essential for embryonic development [78,79].
The ETS1 gene function in tumorigenesis
Using two different highly tumorigenic human colon cancer cell lines DLD-1 and HCT116 that do not express endogenous ETS1 protein, Suzuki, et al. [80] found that ETS1 reversed the transformed phenotype in a dose-dependent fashion, indicating its tumor-suppressive potential [80]. The result is very important since Ets1 bind to WT p53 for tumor suppression (mentioned later in this review). It is a good model for studying suppression of tumorigenicity by ETS1 at transcriptional level, leading to the design and development of novel drugs for cancer therapy [80].
Hashiya et al. expressed human ETS1 into rat hindlimb and found that it stimulated angiogenesis, as measured by increased capillary density and blood flow [81]. Overexpression of ETS1 upregulated the concentrations of hepatocyte growth factor and vascular endothelial growth factor in rat hindlimb. Their study demonstrated that ETS1 regulated angiogenesis through the induction of angiogenic growth factors. Blocking ETS1 expression may thus provide a new therapeutic strategy to treat peripheral arterial disease [81,82].
Alteration of ETS1 in human cancer
The ETS family of transcription factors regulates the expression of genes involved in normal cell development, proliferation, and differentiation [4-6,83-85]. Therefore, the dysregulation of these transcription factors facilitates cell proliferation in many cancers, and several ETS members have been shown to participate in invasion and metastasis by activating gene transcription [83-85]. In three patients with acute monocytic leukemia (AMoL) and t(9;11)(p22;q23) showed that the breakpoint on 9p split the interferon genes and that the Interferon β1 gene was translocated to chromosome 11 [86]. As a consequence, the ETS1 gene was translocated from chromosome 11 to 9p adjacent to interferon genes. They suggested that juxtaposition of interferon and ETS1 genes might be involved in the pathogenesis of AMoL [86].
ETS1 overexpression is also found in human breast cancer (BC) associated with invasiveness and poor prognosis [87,88]. By overexpressing Ets1 or a dominant-negative mutant in BC cells, it was shown that ETS1 plays a key role in coordinating multiple invasive features of cancer cells [87]. They noticed that ETS1 decreased the density of BC cells cultured in three-dimensional extracellular matrix gels. ETS1 overexpression was deleterious to anchorage-independent growth of cells in soft agar. The role of ETS1 was confirmed in vivo, during primary tumor growth and metastatic assay of lung colonization [87] associated with epithelial-to-mesenchymal transition features [89] and high Ki67 [90] - positive cells. siRNA-mediated knockdown of ETS1 in human BC cell lines decreased colony growth, both in anchorage-independent assays and 3D extracellular matrix cultures. These observations suggested an oncogenic role of ETS1 in breast tumorigenesis [87]. In contrast to ETS1, loss of the Myb-like transcription factor DMP1α (DMTF1α) binds to the Ets site in the Arf promoter [91] is associated with low Ki67 and is associated with favorable prognosis [92] for Dmp1 [93-101,67,102-106 for reviews]. Dmp1α also binds directly to p53 for activation in Arf-deficient cells [100,101]. It was reported that expression of neuNT, but not normal neu, caused transcriptional activation of Ets, AP1, or NF-kB-dependent reporter genes [107]. Co-transfection of dominant inhibitory Ets2 mutants specifically blocked neuNT-mediated activation of Ets-dependent reporter genes [107]. Thus, Ets activation is required for neuNT-mediated cellular transformation [105,108-110].
Previous reports have demonstrated that ETS1 is required for activation of the RAS/ERK pathway and migration of RAS/ERK-activated cells by transcriptional activation through ETS/AP1 sites [111,112]. In invasive BC, upregulation of ETS1 was associated with high aggressiveness and poor prognosis. Consistent with this observation, ETS1 regulates the expression of important angiogenetic and extracellular matrix remodeling factors such as VEGF, matrix metalloproteinases [113,114]. Furthermore, ETS1 represses genes such as DUSP4, DUSP6, and SPRY4, all of which provide negative feedback to the RAS/ERK pathway [85]. Statins, small molecule inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoAR), exert anti-tumor effects by altering the RAS/RAF/EK/ERK and RAS/PIK3CA/AKT signaling pathways [115,116]. Lipophilic statins inhibit the growth and proliferation of BC cells, especially hormone receptor-negative, basal-like BC cells [117-120]. Based on these studies, Jung et al. hypothesized that statins suppress TNBC growth by altering the expression of DUSP4 and ETS1 [121]. They demonstrated that ETS1 mRNA and protein were overexpressed in TNBC cells compared with other BC cell lines, where DUSP4 mRNA was downregulated. In addition, simvastatin restored Dusp4-deficiency and suppressed ETS1 expression in TNBC. Moreover they found that depletion of DUSP4 overcame the anti-tumor activity of statins. MAPK pathway inhibitor, U0126 and PI3KCA inhibitor LY294002 also decreased levels of Ets1, phosphor-ERK and phosphor-AKT on Western blot assays. Taken together, these studies suggest the oncogenic function of Ets1 in cancer, and simvastatin potentially affects the activity of transcriptional factors such as ETS1 and DUSP4 through the MAPK pathway [121]. If ETS1 is a critical mediator for the RAS/MEK/ERK signaling, it could act as a tumor promoter or repressor dependent on the biological condition [122-125] because excessive stimulation of this pathway result in Arf-p53 dependent senescence.
Expression of ETS2 in human cancer: influences on Cyclin D1 and p16INK4a expression
One important function of ETS2 is the Cyclin D1 promoter regulation [49,126]. Cyclin D1, the regulatory subunit of several cyclin-dependent kinases (CDK2/4/6), is required for, and capable of shortening, the G1 phase of the cell cycle [127,128]. The transforming mutants of p21Ras (RasVal12, RasLeu61) induces the Cyclin D1 promoter in human trophoblasts, mink lung epithelial, and in Chinese hamster ovary fibroblast cell lines [49]. Site-directed mutagenesis of AP1-like sequences at -954 abolished p21Ras-dependent activation of Cyclin D1 expression. The AP1-like sequences were also required for activation of the Cyclin D1 promoter by c-Jun [49]. In electrophoretic mobility shift assays, several AP1 proteins (Fos/Jun) bound the cyclin D1-954 region. The Cyclin D1 promoter was stimulated by overexpression of mitogen-activated protein kinase (p41MAPK) or c-Ets2 through the proximal 22 base pairs [49]. Conversely, expression of plasmids encoding either dominant negative MAPK (p41MAPKi) or dominant-negatives of ETS activation, antagonized MAPK-dependent induction of Cyclin D1 promoter activity. In summary Ets2 plays critical role in Cyclin D1 induction in response to mitogenic pathways mediated by Ras and MAPK [49,126-130].
The p16INK4a cyclin-dependent kinase inhibitor (CDKN2a) has been implicated in replicative senescence, the state of permanent growth arrest driven by cell divisions or constitutive Ras-Raf-MEK signaling [131-134] (Figure 2). The p16INK4a cyclin-dependent kinase inhibitor is implicated in replicative senescence, the state of permanent growth arrest caused by cumulative cell divisions or constitutive Ras-Raf-MEK signaling [50]. p16INK4a is an essential tumor suppressor that prevents the emergence of incipient cancer cells [134]. The gene expression is regulated at the transcription level and thus the promoter analysis is critical [135,136]. Regulation of p16INK4a expression occurs at the transcriptional level, and involves epigenetic control and multiple transcription factors. PRC1 (Pombe repressor complex 1) and PRC2 (Pombe repressor complex 2) proteins, and histone deacetylases play an important role in the promoter hypermethylation for suppressing p16INK4a expression by YY1 [137] and Id1 [138]. Ohtani, et al. [50] demonstrate a role for the Ets1 and Ets2 transcription factors based on their ability to activate the p16INK4a promoter through the consensus sequences, and their patterns of expression in human diploid fibroblasts. The induction of p16INK4a by Ets2, which was abundant in young human diploid fibroblasts, was potentiated by signaling through the Ras-Raf-MEK kinase cascade, and inhibited by a direct interaction with the helix-loop-helix protein Id1 [50,138] (Figure 1B). In senescent cells, where the ETS2 levels and MEK signaling decline, they saw a marked increase in p16INK4a expression consistent with the reciprocal reduction of Id1 and accumulation of ETS1 [50,138] (Figure 2). These results indicated the opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence.
The tumor suppressor PTEN has both lipid and protein phosphatase activities [139-141]. Its lipid phosphatase activity is essential for its tumor-suppressive function via the phosphoinositide 3-kinase (PI3K) and AKT1 pathways. Weng et al. showed that overexpression of wild type PTEN in the MCF7 breast cancer cells resulted in a phosphatase activity-dependent decrease in the phosphorylation of ETS2 [142]. Interestingly, exposure of MCF7 cells to insulin, insulin-like growth factor-1, or epidermal growth factor lead to the phosphorylation of ETS2. They also reported that the MAP2K1 inhibitor PD590089 cancelled insulin-stimulated phosphorylation of ETS2 [142]. Overexpression of PTEN abrogated activation of the Ras-responsive enhancer, a target of ETS2 action, in a phosphatase-dependent manner [142]. In short, PTEN blocks insulin-stimulated ETS2 phosphorylation through inhibition of the MAP kinase independently of PI3K, which may be mediated through PTEN's protein phosphatase activity [142].
ETS proteins in telomere maintenance
Telomere repeats at chromosomal ends, critical to genome integrity, are maintained through complicated network of proteins and pathways [11,32,33]. A specialized ribonucleic protein, telomerase, maintains telomere homeostasis through repeated addition to counter intrinsic shortcomings of DNA replication that leads to gradual sequence shortening (telomere erosion) in successive mitoses [143,144]. The telomerase is composed of telomerase reverse transcriptase (TERT) subunit and an RNA component [143]. Telomerase, while present in stem cells, is deactivated due to epigenetic silencing of the rate-limiting TERT upon differentiation in most of somatic cells [11]. However, in most cancer cells, telomerase reactivation remains a ubiquitous process and constitutes one of the major hallmarks [143-145]. Discovery of mutations within the core promoter of the TERT gene that create de novo binding sites for ETS transcription factors [32,33] provided a mechanism for cancer-specific telomerase reactivation [11] (Figures 2 and 3). In human cancers, the TERT promoter mutations have been shown to define subsets of patients with poor prognosis with increased transcription of TERT, telomerase reactivation, resulting in increased telomere length. Thus, the TERT promoter mutations have the potential for therapeutic target for cancer [11,32,33].
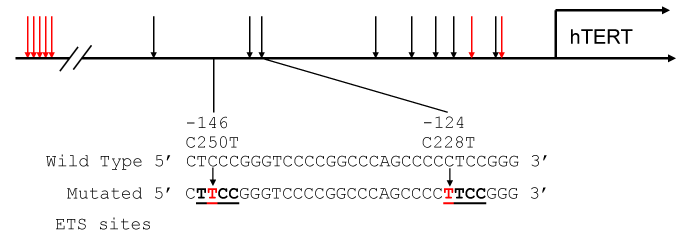
Figure 3. The human TERT promoter [11,143-145]. The proximal promoter has both WT1 (black arrows) and E2F1 (red arrows) -binding sites for regulation. In addition, E2F-binding sites are clustered at 600–1100 base pairs from the transcription initiating G. The hTERT promoter is often mutated to create new ETS sits for transcription factor-binding, which contributes increased telomerase activity in tumor cells.
The Wilms' tumor 1 gene product, which can either be tumor suppressive [146] or oncogenic [147-151], can regulate hTERT gene expression and hence telomerase activity in cancer cells via multiple pathways [12] (Figure 3). It has been reported that WT1 directly bound to p53, and could be either transcriptional repressor or activator dependent on the status of p53 [152,153]. Thus it is likely that WT1 is a transactivator for hTERT rather than repressor in cancer stem cells where p53 in mutated. Indeed, most cases of leukemia show both the TERT and WT1 gene overexpression [154]. Consistently, the TERT and WT1 gene expression analyses provided useful pieces of information for the understanding of chronic myelogenous leukemia progression and their possible usefulness as surrogate markers for treatment monitoring [154].
Ets1/2 and mutant p53
To investigate the basis of its GOF activities of mtp53, Martinez et al. and other labs conducted genome-wide analysis of mtp53 binding, and determined that it could be recruited to promoters through interactions with other transcription factors [44,46,155-164]. Many of the transcription factors that bind to mtp53 have also been shown to interact with wild type p53 (WTp53) (E2F1:DP [104], NF-Y, vitamin D3-responsive element [VDR], and SP1). Early studies showed that mtp53 regulates gene expression via recruitment of transcription factors on the MDR1 promoter [165,166]. Although Sampath study showed that Ets1 interacted with mutant p53, later studies found specific interaction of Ets1 with WTp53 [167,168] which has become a generalized understanding.
Data from ChiP [99], Chip on Chip [169], and Chip-Seq [170] revealed that ~50% of promoters occupied by mtp53 contained ETS-binding sites, suggesting that physical binding with ETS proteins is an essential mechanism by which mtp53 regulates gene expression for oncogenic transformation [159,160]. Importantly, these mtp53-bound genomic regions do not have a WTp53 response element 5′-PuPuPuC(A/T)(A/T)GPyPyPy-3 [171], indicating that the mtp53 proteins do not cause transformation through direct binding to DNA [159]. Although the ETS1 might be important for the regulation of some mtp53 target genes [160], side-by-side comparison using recombinant proteins revealed that mtp53 preferentially associates with ETS2 [44,158]. All structural and DNA contact p53 mutants bound to ETS2 [44,158,162]. Whereas ETS1 knockdown had no effect on mtp53 target gene expression, ETS2 depletion reproduced changes in gene expression that occur on mtp53 depletion suggesting close relationship between mtp53 and ETS2. The observation that ETS2 interacts with various mtp53 (R175H, R248Q, R248W, R249S, R273H, R273L, and R280K) suggests that through ETS2 binding, different mtp53 proteins reveal oncogenic potential through common mechanisms consistent with the recent MD Anderson study [172].
The VCU group studied the mechanism for mtp53 binding by mapping the human genomic binding sites for p53R273H (one of GOF mtp53) using ChIP-seq and showed that the protein was localized to ETS DNA sequence motifs with ETS1 and GABPA binding, both within promoters and distal to promoters [160]. p53R273H showed significant and substantial binding to bidirectional promoters, which were enriched for inverted repeated ETS DNA sequence motifs [160]. p53R273H showed an exponential increase in probability of binding to promoters with a higher number of ETS motifs [160]. The same group reported that in cells expressing p53R273H GOF mtp53 simultaneously upregulates genes from multiple signaling pathways by recognizing promoters containing distinct transcription factor binding sites [161]. Thus, inhibition of one GOF mtp53-induced signaling cascade would be insufficient to inhibit tumor growth, but rather global inhibition of GOF mtp53 activity in necessary to fond a novel anti-cancer therapeutic agents [161].
It should be remembered that ETS2 binds to the tetramerization domain of p53 [173-175], which is thought to be functionally intact in both WTp53 and mtp53. Then the question is how ETS2 distinguishes between mtp53 and WTp53. One possibility is that p53 mutants adopt a different structure from normal p53 that enables their interaction with ETS2. However, some GOF p53 proteins have similar structures to normal p53 which raises the question for this. The 2nd possibility is when WTp53 in searching for DNA sequences throughout the genome, it might not be able to interact with ETS2 [175]. When WTp53 is bound to genomic DNA, it might alter the structure in a way that not compatible with binding to ETS2 [175]. The 3rd possibility is that the expression of some WTp53 target genes prevents the normal protein interactions found in mtp53 [175]. Whatever the scenario is, it is of paramount importance to elucidate the mechanisms of activation/stabilization of mt53 since it is mutated in 50% of human cancers associated with malignant potential of tumors cells, hence worse prognosis of patients.
Very recently, the Lozano’s group elucidated how GOF mtp53 drove cancer metastasis by developing a traceable osteosarcoma mouse model [172]. They showed that mtp53 mice developed osteosarcomas with increased metastasis as compared with p53-null mice. The RNA-seq analysis of tumors identified a cluster of small nucleolar RNAs (snoRNAs) that were highly up-regulated in mtp53 mutant tumors with enrichment of Ets2 transcription factor-binding site [172]. Consistently, homozygous deletion of Ets2 in p53 mutant mice resulted in significant down-regulation of snoRNAs with simultaneous reversion of the pro-metastatic phenotype of tumors induced my mtp53, which had no effect on the growth of primary osteosarcoma. Thus Ets2 inhibition is a potential therapeutic vulnerability in mtp53 mutant osteosarcomas [172].
Future directions
Ets1 shows both oncogenic and tumor suppressive activity dependent on the assay conditions. It is generally considered an oncogene by aberrant overexpression in human cancers. The role of Ets1 in embryonic maintenance or inhibition of cell proliferation for lymphoid cells is clear from gene knockout studies. Accumulating pieces of evidence has suggested that Ets1 accelerates angiogenesis, and has an oncogenic role mediated by MAPK signaling. Conversely, Ets2 has a tumor-suppressive role by upregulation the transcription of p16INK4a although it stimulates the transcription of Cyclin D1. The situation is completely reversed when the cells have mutant p53; ETS2 reveals oncogenic function by stabilizing mutant p53 as evidenced by the recent study employing gene engineered mice [172]. The authors concluded that blocking Ets2 activity is essential to prevent lung metastasis of osteosarcoma. Thus the search for ETS2 blocker is of paramount importance to prevent cancer metastasis. Both ETS1 and ETS2 stimulate p53; however, neither ETS1 nor ETS2 is in the list of target genes for p53 [176,177]. Currently, the role of other ETS family proteins on activation/inhibition of mutant p53 is unknown. Thus extensive studies should be done in this direction since p53 is mutated in half of human cancers with GOF functions and that ETS have so many other family members.
It is very intriguing to know that human telomerase promoter have novel ETS sites when they are mutated [11]. That may be the reason why many human cancers have increased telomerase activity with long telomeres where the inhibitors are in clinical trials to cancer. Since human TERT promoter has both E2F1 and WT1 binding sites, it will be important how these transcription factors regulate the hTERT promoter. It will be also essential to analyze which ETS proteins bind to the hTERT promoter to translate the findings of ETS mutation to clinical levels.
Declarations
Acknowledgements: We thank all other members of Dr. Inoue’s lab for sharing unpublished research data.
Financial support: K. Inoue was supported by NIH/NCI 2R01CA106314, ACS RSG-07-207-01-MGO, and KG080179.
Conflicts of interest: The authors declare no conflicts of interest.
References
- Bister K, Nunn M, Moscovici C, Perbal B, Baluda M, et al. (1982) Acute leukemia viruses E26 and avian myeloblastosis virus have related transformation-specific RNA sequences but different genetic structures, gene products, and oncogenic properties. Proc Natl Acad Sci USA 79: 3677-3681.
- Moelling K, Pfaff E, Beug H, Beimling P, Bunte T, et al. (1985) DNA-binding activity is associated with purified myb proteins from AMV and E26 viruses and is temperature-sensitive for E26 ts mutants. Cell 40: 983-990.
- Metz T, Graf T (1991) Fusion of the nuclear oncoproteins v-Myb and v-Ets is required for the leukemogenicity of E26 virus. Cell 66: 95-105. [Crossref]
- Findlay VJ, LaRue AC, Turner DP, Watson PM, Watson DK (2013) Understanding the role of ETS-mediated gene regulation in complex biological processes. Adv Cancer Res 119: 1-61. [Crossref]
- Feldman RJ, Sementchenko VI, Watson DK (2003a) The epithelial-specific Ets factors occupy a unique position in defining epithelial proliferation, differentiation and carcinogenesis. Anticancer Res 23: 2125-2131.
- Feldman RJ, Sementchenko VI, Gayed M, Fraig MM, Watson DK (2003b) Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res 63: 4626-4631.
- Hirai H, Sherr CJ (1996) Interaction of D-type cyclins with a novel myb-like transcription factor, DMP1. Mol Cell Biol 16: 6457-6467.
- Shaikhibrahim Z, Wernert N (2012) ETS transcription factors and prostate cancer: the role of the family prototype ETS-1 (review). Int J Oncol 40: 1748-1754. [Crossref]
- Garrett-Sinha LA (2013) Review of Ets1 structure, function, and roles in immunity. Cell Mol Life Sci 70: 3375-3390.
- Dittmer J (2015) The role of the transcription factor Ets1 in carcinoma. Semin Cancer Biol 35: 20-38.
- Heidenreich B, Kumar R (2017) TERT promoter mutations in telomere biology. Mutat Res 771: 15-31. [Crossref]
- Sitaram RT, Degerman S, Ljungberg B, Andersson E, Oji Y, et al. (2010) Wilms' tumour 1 can suppress hTERT gene expression and telomerase activity in clear cell renal cell carcinoma via multiple pathways. Br J Cancer 103: 1255-1262.
- Sizemore GM, Pitarr2021 Copyright OAT. All rights reserv(2017) The ETS family of oncogenic transcription factors in solid tumours. Nat Rev Cancer 17: 337-351
- Yang BS, Hauser CA, Henkel G, Colman MS, Van Beveren C, et al. (1996) Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol 16: 538-547.
- Li R, Pei H, Watson DK (2000) Regulation of Ets function by protein - protein interactions. Oncogene 19: 6514-6523.
- Oikawa T (2004) ETS transcription factors: possible targets for cancer therapy. Cancer Sci 95: 626-633. [Crossref]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K (2005) Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol 25: 220-232.
- Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, et al. (2007) Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 12: 381-394.
- Kabbout M, Dakhlallah D, Sharma S, Bronisz A, Srinivasan R, Piper M, et al. (2014) MicroRNA 17-92 cluster mediates ETS1 and ETS2-dependent RAS-oncogenic transformation. PLoS One 9: e100693.
- Mackereth CD, Schärpf M, Gentile LN, MacIntosh SE, Slupsky CM, et al. (2004) Diversity in structure and function of the Ets family PNT domains. J Mol Biol 342: 1249-1264.
- Langer SJ, Bortner DM, Roussel MF, Sherr CJ, Ostrowski MC (1992) Mitogenic signaling by colony-stimulating factor 1 and ras is suppressed by the ets-2 DNA-binding domain and restored by myc overexpression. Mol Cell Biol 12: 5355-5362.
- Wasylyk C, Maira SM, Sobieszczuk P, Wasylyk B (1994) Reversion of Ras transformed cells by Ets transdominant mutants. Oncogene 9: 3665-3673.
- Hever A, Oshima RG, Hauser CA (2003) Ets2 is not required for Ras or Neu/ErbB-2 mediated cellular transformation in vitro. Exp Cell Res 290: 132-143. [Crossref]
- Sizemore GM, Pitarresi JR, Balakrishnan S, Ostrowski MC (2017) The ETS family of oncogenic transcription factors in solid tumours. Nat Rev Cancer 17: 337-351. [Crossref]
- Sacchi N, Watson DK, Guerts van Kessel AH, Hagemeijer A, Kersey J, et al. (1986) Hu-ets-1 and Hu-ets-2 genes are transposed in acute leukemias with (4;11) and (8;21) translocations. Science 231: 379-382. [Crossref]
- Sacchi N, Cheng SV, Tanzi RE, Gusella JF, Drabkin HA, et al. (1988) The ETS genes on chromosome 21 are distal to the breakpoint of the acute myelogenous leukemia translocation (8;21). Genomics 3: 110-116.
- Nucifora G, Birn DJ, Erickson P, Gao J, LeBeau MM, et al. (1993) Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood 81: 883-888.
- Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, et al. The (8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J 12: 2715-2721.
- Rao VN, Papas TS, Reddy ESP (1987) ERG, a human ETS-related gene on chromosome 21: alternative splicing, polyadenylation, and translation. Science 237: 635-639.
- Rao VN, Modi WS, Drabkin HD, Patterson D, O'Brien SJ, et al. (1988) The human ERG gene maps to chromosome 21, band q22: relationship to the 8;21 translocation of acute myelogenous leukemia. Oncogene 3: 497-500.
- Murakami K, Mavrothalassitis G, Bhat NK, Fisher RJ, Papas TS (1993) Human ERG-2 protein is a phosphorylated DNA-binding protein--a distinct member of the ETS family. Oncogene 8: 1559-1566.
- Heidenreich B, Rachakonda PS, Hemminki K, Kumar R (2014) TERT promoter mutations in cancer development. Curr Opin Genet Dev 24: 30-37.
- Heidenreich B, Nagore E, Rachakonda PS, Garcia-Casado Z, Requena C, et al. (2014) Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat Commun 5: 3401.
- Avin BA, Umbricht CB, Zeiger MA (2016) Human telomerase reverse transcriptase regulation by DNA methylation, transcription factor binding and alternative splicing. Int J Oncol 49: 2199-2205.
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339: 957-959.
- Quaas A, Oldopp T, Tharun L, Klingenfeld C, Krech T, et al. (2014) Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch 465: 673-677.
- Dwyer J, Li H, Xu D, Liu JP (2007) Transcriptional regulation of telomerase activity: roles of the (sic) Ets transcription factor family. Ann New York Acad Sci 1114: 36-47.
- Parrales A, Iwakuma T (2015) Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol 5: 288. [Crossref]
- Vaughan CA, Singh S, Windle B, Sankala HM, Graves PR, et al. (2012) p53 mutants induce transcription of NF-κB2 in H1299 cells through CBP and STAT binding on the NF-κB2 promoter and gain of function activity. Arch Biochem Biophys 518: 79-88.
- Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, et al. (2016) Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol 18: 897-909.
- Frazier MW, He X, Wang J, Gu Z, Cleveland JL, et al. (1998) Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol 18: 3735-3743. [Crossref]
- Liao P, Zeng SX, Zhou X, Chen T, Zhou F, et al. (2017) Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol Cell 68:1134-1146.e6.
- Ferraiuolo M, Di Agostino S, Blandino G, Strano S. (2016) Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front Oncol 6: 77.
- Martinez LA (2016) Mutant p53 and ETS2, a Tale of Reciprocity. Front Oncol 6: 35.
- Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, et al. (2015) Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525: 206-211. [Crossref]
- Vaughan C, Pearsall I, Yeudall A, Deb SP, Deb S (2014) p53: its mutations and their impact on transcription. Subcell Biochem 85: 71-90.
- Kühn MW, Bullinger L, Gröschel S, Krönke J, Edelmann J, et al. (2014) Genome-wide genotyping of acute myeloid leukemia with translocation t(9;11)(p22;q23) reveals novel recurrent genomic alterations. Haematologica 99: e133-e135.
- Léglise MC, Rivière D, Brière J (1990) Acute Leukemia with a Translocation T(4;11)(q21;q23): a Distinct Clinicopathological Entity: Report of a Case with Cytogenetic Clonal Evolution and Review of 146 Cases of the Literature. Leuk Lymphoma 2: 353-368.
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, et al. (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 270: 23589-23597.
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, et al. (2001) Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 409: 1067-1070.
- Dwyer JM, Liu JP (2010) Ets2 transcription factor, telomerase activity and breast cancer. Clin Exp Pharmacol Physiol 37: 83-87.
- Watson DK, McWilliams MJ, Lapis P, Lautenberger JA, Schweinfest CW, et al. (1988) Mammalian ets-1 and ets-2 genes encode highly conserved proteins. Proc Natl Acad Sci USA 85: 7862-7866.
- de Taisne C, Gegonne A, Stehelin D, Bernheim A, Berger R (1984) Chromosomal localization of the human proto-oncogene c-ets. Nature 310: 581-583. [Crossref]
- Watson DK, McWilliams-Smith MJ, Nunn MF, Duesberg PH, O'Brien SJ, et al. (1985) The ets sequence from the transforming gene of avian erythroblastosis virus, E26, has unique domains on human chromosomes 11 and 21: both loci are transcriptionally active. Proc Natl Acad Sci USA 82: 7294-7298.
- Fujiwara S, Fisher RJ, Seth A, Bhat NK, Showalter SD, et al. (1988) Characterization and localization of the products of the human homologs of the v-ets oncogene. Oncogene 2: 99-103.
- Nelson ML, Kang HS, Lee GM, Blaszczak AG, Lau DK, et al. (2010) Ras signaling requires dynamic properties of Ets1 for phosphorylation-enhanced binding to coactivator CBP. Proc Natl Acad Sci USA 107: 10026-10031.
- Laitem C, Leprivier G, Choul-Li S, Begue A, Monte D, et al. (2009) Ets-1 p27: a novel Ets-1 isoform with dominant-negative effects on the transcriptional properties and the subcellular localization of Ets-1 p51. Oncogene 28: 2087-2099.
- Mavrothalassitis GJ, Watson DK, Papas TS (1990) Molecular and functional characterization of the promoter of ETS2, the human c-ets-2 gene. Proc Natl Acad Sci USA 87: 1047-1051.
- Owczarek CM, Portbury KJ, Hardy MP, O'Leary DA, Kudoh J, et al. (2004) Detailed mapping of the ERG-ETS2 interval of human chromosome 21 and comparison with the region of conserved synteny on mouse chromosome 16. Gene 324: 65-77.
- Basuyaux JP, Ferreira E, Stéhelin D, Butticè G (1997) The Ets transcription factors interact with each other and with the c-Fos/c-Jun complex via distinct protein domains in a DNA-dependent and –independent manner. J Biol Chem 272: 26188-26195.
- Eyquem S, Chemin K, Fasseu M, Bories JC (2004) The Ets-1 transcription factor is required for complete pre-T cell receptor function and allelic exclusion at the T cell receptor beta locus. Proc Natl Acad Sci USA 101: 15712-15717.
- Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, et al. (2005) Ets-1 is a critical regulator of Ang II-medicated vascular inflammation and remodeling. J Clin Invest 115: 2508-2516.
- Maglic D, Stovall DB, Cline JM, Fry EA, Mallakin A, et al. (2015) DMP1β, a splice isoform of the tumor suppressor DMP1 locus, induces proliferation and progression of breast cancer. J Pathol 236: 90-102.
- Inoue K, Fry EA (2015) Aberrant Splicing of Estrogen Receptor, HER2, and CD44 Genes in Breast Cancer. Genet Epigenet 7: 19-32.
- Inoue K, Fry EA (2014) Alterations of p63 and p73 in human cancers in ‘Mutant p53 and MDM2 in cancer’. Editors: Swati Palit Deb & Sumitra Deb. Springer, NY. Chapter 2: 17-40, 2014. Subcell Biochem 85: 17-40.
- Inoue K, Fry EA (2016) Novel Molecular Markers for Breast Cancer. Biomark Cancer 8: 25-42. [Crossref]
- Inoue K, Fry EA (2016) Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int J Cancer 139: 33-41.
- Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI (2016) Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35: 2413-2427.
- Abou Faycal C, Gazzeri S, Eymin B (2016) RNA splicing, cell signaling, and response to therapies. Curr Opin Oncol 28: 58-64. [Crossref]
- Luz FA, Brígido PC, Moraes AS, Silva MJ (2017) Aberrant Splicing in Cancer: Mediators of Malignant Progression through an Imperfect Splice Program Shift. Oncology 92: 3-13.
- Jiang Q, Crews LA, Holm F, Jamieson CHM (2017) RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat Rev Cancer 17: 381-339.
- Higuchi T, Bartel FO, Masuya M, Deguchi T, Henderson KW, et al. (2007) Thymomegaly, microsplenia, and defective homeostatic proliferation of peripheral lymphocytes in p51-Ets1 isoform-specific null mice. Mol Cell Biol 27: 3353-366.
- Bartel FO, Higuchi T, Spyropoulos DD (2000) Mouse models in the study of the Ets family of transcription factors. Oncogene 19: 6443-6454.
- Sumarsono SH, Wilson TJ, Tymms MJ, Venter DJ, Corrick CM, et al. (1996) Down's syndrome-like skeletal abnormalities in Ets2 transgenic mice. Nature 379: 534-537. [Crossref]
- Wolvetang EJ, Wilson TJ, Sanij E, Busciglio J, Hatzistavrou T, et al. (2003) ETS2 overexpression in transgenic models and in Down syndrome predisposes to apoptosis via the p53 pathway. Hum Mol Genet 12: 247-255.
- Yamamoto H, Flannery ML, Kupriyanov S, Pearce J, McKercher SR, et al. (1998) Defective trophoblast function in mice with a targeted mutation of Ets2. Genes Dev 12: 1315-1326.
- Georgiades P, Rossant J (2006) Ets2 is necessary in trophoblast for normal embryonic anteroposterior axis development. Development 133:1059-1068.
- Wen F, Tynan JA, Cecena G, Williams R, Múnera J, et al. (2007) Ets2 is required for trophoblast stem cell self-renewal. Dev Biol 312: 284-299. [Crossref]
- Tallquist MD, Soriano P (2000) Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26: 113-115.
- Suzuki H, Romano-Spica V, Papas TS, Bhat NK (1995) ETS1 suppresses tumorigenicity of human colon cancer cells. Proc Natl Acad Sci USA 92: 4442-4446.
- Hashiya N, Jo N, Aoki M, Matsumoto K, Nakamura T, et al. (2004) In vivo evidence of angiogenesis induced by transcription factor Ets-1: Ets-1 is located upstream of angiogenesis cascade. Circulation 109: 3035-3041.
- Lincoln DW 2nd, Bove K (2005) The transcription factor Ets-1 in breast cancer. Front Biosci 10: 506-511. [Crossref]
- Hahne JC, Okuducu AF, Sahin A, Fafeur V, Kiriakidis S, et al. (2008) The transcription factor ETS-1: its role in tumour development and strategies for its inhibition. Mini Rev Med Chem 8: 1095-1105.
- Turner DP, Watson DK (2008) ETS transcription factors: oncogenes and tumor suppressor genes as therapeutic targets for prostate cancer. Expert Rev Anticancer Ther 8: 33-42.
- Kar A, Gutierrez-Hartmann A (2013) Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit Rev Biochem Mol Biol 48: 522-543.
- Diaz MO, Le Beau MM, Pitha P, Rowley JD (1986) Interferon and c-ets-1 genes in the translocation (9;11)(p22;q23) in human acute monocytic leukemia. Science 231: 265-267. [Crossref]
- Furlan A, Vercamer C, Bouali F, Damour I, Chotteau-Lelievre A, et al. (2014) Ets-1 controls breast cancer cell balance between invasion and growth. Int J Cancer 135: 2317-2328.
- Dittmer J (2015) The role of the transcription factor Ets1 in carcinoma. Semin Cancer Biol 35: 20-38. [Crossref]
- Mittal V (2018) Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol 13: 395-412.
- Taneja P, Maglic D, Kai F, Zhu S, Kendig RD et al. (2010) Classical and novel molecular prognostic markers for human breast cancer and their clinical significance. Clin Med Oncol 4: 15-34.
- Inoue K, Roussel MF, Sherr CJ (1999) Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci U S A 96: 3993-3998. [Crossref]
- Maglic D, Zhu S, Fry EA, Taneja P, Kai F, et al. (2013) Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene 32: 4120-4129.
- Inoue K, Sherr CJ (1998) Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol 18: 1590-1600.
- Inoue K, Sherr CJ, Shapiro LH (1998) Regulation of the CD13/Aminopeptidase N gene by DMP1, a transcription factor antagonized by D-type cyclins. J Biol Chem 273: 29188-29194.
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, et al. (2000) Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, ras transformation, and tumorigenesis. Genes Dev 14: 1797-1809.
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ (2001) Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev 15: 2934-2939.
- Mallakin A, Taneja P, Matise, LA, Willingham MC, Inoue K (2006) Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene 25: 7703-7713.
- Taneja P, Mallakin A, Matise LA, Frazier DP, Choudhary M, et al. (2007) Repression of Dmp1 and Arf 98. transcription by anthracyclins: critical roles of the NF-kappaB subunit p65. Oncogene 26: 7457-7466. [Crossref]
- Mallakin A, Sugiyama T, Kai F, Taneja P, Kendig RD, et al. (2010) The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer 126: 1403-1416.
- Frazier DP, Kendig RD, Kai F, Maglic D, Sugiyama T, et al. (2012) Dmp1 physically interacts with p53 and positively regulates p53’s stabilization, nuclear localization, and function. Cancer Res 72: 1740-1750.
- Kendig RD, Kai F, Fry EA, Inoue K (2017) Stabilization of the p53-DNA Complex by the Nuclear Protein Dmp1α. Cancer Invest 35: 301-312. [Crossref]
- Inoue K, Mallakin A, Frazier DP (2007) Dmp1 and tumor suppression. Oncogene 26: 4329-4335.
- Inoue K, Fry EA, Frazier DP (2016) Transcription factors that interact with p53 and Mdm2. Int J Cancer 138: 1577-1585. [Crossref]
- Fry EA, Taneja P, Inoue K (2017) Oncogenic and tumor-suppressive mouse models for breast cancer employing HER2/neu. Int J Cancer 140: 495-503.
- Inoue K, Fry EA (2017) Haplo-insufficient tumor suppressor genes. A book chapter in ‘Advances in Medicine and Biology’. Nova Science Publishers, Inc. 118: 83-122.
- Inoue K, Fry EA (2018) Aberrant Expression of ARF in human cancer - a new biomarker? Tumor and Microenvironment, in press.
- Galang CK, García-Ramírez J, Solski PA, Westwick JK, Der CJ, et al. (1996) Oncogenic Neu/ErbB-2 increases ets, AP-1, and NF-kappaB-dependent gene expression, and inhibiting ets activation blocks Neu-mediated cellular transformation. J Biol Chem 271: 7992-7998.
- Taneja P, Maglic D, Kai F, Sugiyama T, Kendig RD, et al. (2010) Critical roles of DMP1 in human epidermal growth factor receptor 2/neu-Arf-p53 signaling and breast cancer development. Cancer Res 70: 9084-9094.
- Fry EA, Taneja P, Maglic D, Zhu S, Sui G, et al. (2013) Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One 8: e77870. [Crossref]
- Fry EA, Taneja P, Inoue K (2016) Clinical applications of mouse models for breast cancer engaging HER2/neu. Integr Cancer Sci Ther 3: 593-603. [Crossref]
- Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, et al. (2011) Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev 25: 2147-2157. [Crossref]
- Selvaraj N, Budka JA, Ferris MW, Plotnik JP, Hollenhorst PC (2015) Extracellular signal-regulated kinase signaling regulates the opposing roles of JUN family transcription factors at ETS/AP-1 sites and in cell migration. Mol Cell Biol 35: 88-100.
- Davidson B, Goldberg I, Gotlieb WH, Kopolovic J, Risberg B, et al. (2003) Coordinated expression of integrin subunits, matrix metalloproteinases (MMP), angiogenic genes and Ets transcription factors in advanced-stage ovarian carcinoma: a possible activation pathway? Cancer Metastasis Rev 22: 103-115.
- Ghosh S, Basu M, Roy SS (2012) ETS-1 protein regulates vascular endothelial growth factor-induced matrix metalloproteinase-9 and matrix metalloproteinase-13 expression in human ovarian carcinoma cell line SKOV-3. J Biol Chem 287: 15001-15015.
- Yanae M, Tsubaki M, Satou T, Itoh T, Imano M, et al. (2011) Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J Exp Clin Cancer Res 30: 74.
- Tsubaki M, Yamazoe Y, Yanae M, Satou T, Itoh T, et al. (2011) Blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathways by statins reduces the expression of bFGF, HGF, and TGF-β as angiogenic factors in mouse osteosarcoma. Cytokine 54: 100-107.
- Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, et al. (2006) Breast cancer growth prevention by statins. Cancer Res 66: 8707-8714.
- Garwood ER, Kumar AS, Baehner FL, Moore DH, Au A, et al. (2010) Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res Treat 119: 137-144.
- Goard CA, Chan-Seng-Yue M, Mullen PJ, Quiroga AD, Wasylishen AR, et al. (2014) Identifying molecular features that distinguish fluvastatin-sensitive breast tumor cells. Breast Cancer Res Treat 143: 301-312.
- Vallianou NG, Kostantinou A, Kougias M, Kazazis C1 (2014) Statins and cancer. Anticancer Agents Med Chem 14: 706-712. [Crossref]
- Jung HH, Lee SH, Kim JY, Ahn JS, Park YH, et al. (2016) Statins affect ETS1-overexpressing triple-negative breast cancer cells by restoring DUSP4 deficiency. Sci Rep 6: 33035.
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593-602.
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, et al. (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12: 3008-3019.
- Lin AW, Lowe SW (2001) Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci U S A 98: 5025-5030. [Crossref]
- de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, et al. (2004) PML is a direct p53 target that modulates p53 effector functions. Mol Cell 13: 523-535. [Crossref]
- Carbone GM, Napoli S, Valentini A, Cavalli F, Watson DK et al. (2004) Triplex DNA-mediated downregulation of Ets2 expression results in growth inhibition and apoptosis in human prostate cancer cells. Nucleic Acids Res 32: 4358-4367.
- Zhu S, Mott RT, Fry EA, Taneja P, Kulik G, et al. (2013) Cooperation between Dmp1 loss and cyclin D1 overexpression in breast cancer. Am J Pathol 183: 1339-1350. [Crossref]
- Inoue K, Fry EA (2015) Aberrant expression of Cyclin D1 in cancer. Signal Transduction Insights 4: 1-13.
- Li Y, Chitnis N, Nakagawa H, Kita Y, Natsugoe S, et al. (2015) PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov 5: 288-303.
- Qie S, Diehl JA (2016) Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl) 94: 1313-1326. [Crossref]
- Svensson Månsson S, Reis-Filho J, Landberg G (2007) Transcriptional upregulation and unmethylation of the promoter region of p16 in invasive basal cell carcinoma cells and partial co-localization with the gamma 2 chain of laminin-332. J Pathol 212: 102-111.
- Rayess H, Wang MB, Srivatsan ES (2012) Cellular senescence and tumor suppressor gene p16. Int J Cancer 130: 1715-1725.
- Sharpless NE, Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15: 397-408.
- Inoue K, Fry EA (2018) Expression of p16INK4a in human cancer - a new biomarker? Cancer Reports and Reviews 2: 1-7.
- Hara E, Smith R, Parry D, Tahara H, Stone S, et al. (1996) Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 16: 859-867. [Crossref]
- Yogev O, Anzi S, Inoue K, Shaulian E (2006) Induction of transcriptionally active Jun proteins regulates drug-induced senescence. J Biol Chem 281: 34475-34483.
- Zhang Q, Stovall DB, Inoue K, Sui G (2011) The Oncogenic Role of Yin Yang 1. Crit Rev Oncog 16: 163-197.
- Zebedee Z, Hara E (2001) Id proteins in cell cycle control and cellular senescence. Oncogene 20: 8317-8325. [Crossref]
- Berger AH, Pandolfi PP (2011) Haplo-insufficiency: a driving force in cancer. J Pathol 223: 137-146.
- Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13: 283-296. [Crossref]
- Inoue K, Fry EA, Taneja P (2013) Recent progress in mouse models for tumor suppressor genes and its implications in human cancer. Clin Med Oncol 7: 103-122.
- Weng L-P, Brown JL, Baker K., Ostrowski MC, Eng C (2002) PTEN blocks insulin-mediated ETS-2 phosphorylation through MAP kinase, independently of the phosphoinositide 3-kinase pathway. Hum Mol Genet 11: 1687-1696.
- Xu L, Li S, Stohr BA (2013) The role of telomere biology in cancer. Annu Rev Pathol 8: 49-78. [Crossref]
- Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 350: 1193-1198.
- Lu W, Zhang Y, Liu D, Songyang Z, Wan M (2013) Telomeres-structure, function, and regulation. Exp Cell Res 319: 133-141. [Crossref]
- Shandilya J, Roberts SG (2015) A role of WT1 in cell division and genomic stability. Cell Cycle 14: 1358-1364.
- Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, et al. (1994) WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 84: 3071-3079.
- Yamagami T, Sugiyama H, Inoue K, Ogawa H, Tatekawa T, et al. (1996) Growth inhibition of human leukemic cells by WT1 antisense oligonucleotides. Blood 87: 2878-2884.
- Tamaki H, Ogawa H, Inoue K, Soma T, Yamagami T, et al. (1996) Increased expression of the Wilms tumor gene (WT1) at relapse in acute leukemia. Blood 88: 4396-4398. [Crossref]
- Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, et al. (1997) Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood 89: 1405-1412.
- Inoue K, Tamaki H, Ogawa H, Oka Y, Soma T, et al. (1998). Wilms’ tumor gene (WT1) competes with differentiation-inducing signal in hematopoietic progenitor cells. Blood 91: 2969-2976.
- Maheswaran S, Park S, Bernard A, Morris JF, Rauscher FJ 3rd, et al. (1993) Physical and functional interaction between WT1 and p53 proteins. Proc Natl Acad Sci USA 90: 5100-5104.
- Maheswaran S, Englert C, Bennett P, Heinrich G, Haber DA (1995) The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev 9: 2143-2156.
- Varma N, Anand MS, Varma S, Juneja SS (2011) Role of hTERT and WT1 gene expression in disease progression and imatinib responsiveness of patients with BCR-ABL positive chronic myeloid leukemia. Leuk Lymphoma 52: 687-693.
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, et al. (2006) Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10: 191-202.
- Fontemaggi G, Dell'Orso S, Trisciuoglio D, Shay T, Melucci E, et al. (2009) The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol 16: 1086-1093. [Crossref]
- Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, et al. (2010) Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 17: 273-285.
- Do PM, Varanasi L, Fan S, Li C, Kubacka I, et al. (2012) Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev 26: 830-845.
- Yeudall WA, Wrighton KH, Deb S (2013) Mutant p53 in cell adhesion and motility. Methods Mol Biol 962: 135-146. [Crossref]
- Vaughan CA, Deb SP, Deb S, Windle B (2014) Preferred binding of gain-of-function mutant p53 to bidirectional promoters with coordinated binding of ETS1 and GABPA to multiple binding sites. Oncotarget 5: 417-427.
- Vaughan CA, Singh S, Grossman SR, Windle B, Deb SP, et al. (2017) Gain-of-function p53 activates multiple signaling pathways to induce oncogenicity in lung cancer cells. Mol Oncol 11: 696-711.
- Liu DD, Kang Y (2017) Ets2 anchors the prometastatic function of mutant p53 in osteosarcoma. Genes Dev 31: 1823-1824. [Crossref]
- Liao P, Zeng SX, Zhou X, Chen T, Zhou F, et al. (2017) Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol Cell 68: 1134-1146.e6.
- Tanaka N, Zhao M, Tang L, Patel AA, Xi Q, et al. (2018) Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene 37: 1279-1292.
- Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, et al. (2001) Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 276: 39359-39367.
- Deb D, Scian M, Roth KE, Li W, Keiger J, et al. (2002) Hetero-oligomerization does not compromise 'gain of function' of tumor-derived p53 mutants. Oncogene 21: 176-189.
- Kim E, Günther W, Yoshizato K, Meissner H, Zapf S, et al. (2003) Tumor suppressor p53 inhibits transcriptional activation of invasion gene thromboxane synthase mediated by the proto-oncogenic factor ets-1. Oncogene 22: 7716-7727.
- Xu D, Wilson TJ, Chan D, De Luca E, Zhou J, et al. (2002) Ets1 is required for p53 transcriptional activity in UV-induced apoptosis in embryonic stem cells. EMBO J 21: 4081-4093.
- Goeman F, Fontemaggi G, Blandino G (2013) ChIP-on-chip to identify mutant p53 targets. Methods Mol Biol 962: 211-226. [Crossref]
- Garritano S, Inga A, Gemignani F, Landi S (2013) More targets, more pathways and more clues for mutant p53. Oncogenesis 2: e54.
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1: 45-49.
- Pourebrahim R, Zhang Y, Liu B, Gao R, Xiong S, et al. (2017) Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev 31: 1847-1857.
- Beckerman R, Yoh K, Mattia-Sansobrino M, Zupnick A, Laptenko O, et al. (2016) Lysines in the tetramerization domain of p53 selectively modulate G1 arrest. Cell Cycle 15: 1425-1438.
- Kamada R, Toguchi Y, Nomura T, Imagawa T, Sakaguchi K (2016) Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 106: 598-612. [Crossref]
- Prives C, Lowe SW (2015) Cancer: Mutant p53 and chromatin regulation. Nature 525: 199-200.
- Beckerman R, Prives C (2010) Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2: a000935. [Crossref]
- Fischer M (2017) Census and evaluation of p53 target genes. Oncogene 36: 3943-3956.
- Le Gallic L, Virgilio L, Cohen P, Biteau B, Mavrothalassitis G (2004) ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression. Mol Cell Biol 24: 1206-1218.
- Okamura K, Yamashita S, Ando H, Horibata Y, Aoyama C, et al. (2009) Identification of nuclear localization and nuclear export signals in Ets2, and the transcriptional regulation of Ets2 and CTP:phosphocholine cytidylyltransferase alpha in tetradecanoyl-13-acetate or macrophage-colony stimulating factor stimulated RAW264 cells. Biochim Biophys Acta 1791: 173-182.
- Nagulapalli S, Atchison ML (1998) Transcription factor Pip can enhance DNA binding by E47, leading to transcriptional synergy involving multiple protein domains. Mol Cell Biol 18: 4639-4650.
- Sharrocks AD1 (2001) The ETS-domain transcription factor family. Nat Rev Mol Cell Biol 2: 827-837. [Crossref]
- Myers E, Hill AD, Kelly G, McDermott EW, O'Higgins NJ, et al. (2005) Associations and interactions between Ets-1 and Ets-2 and coregulatory proteins, SRC-1, AIB1, and NCoR in breast cancer. Clin Cancer Res 11: 2111-2122.
- Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, et al. (2009) Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev 23: 681-693. [Crossref]
- Gallant S, Gilkeson G (2006) ETS transcription factors and regulation of immunity. Arch Immunol Ther Exp (Warsz) 54: 149-163. [Crossref]
- Tetsu O, McCormick F (2017) ETS-targeted therapy: can it substitute for MEK inhibitors? Clin Transl Med 6: 16.