Key Words
inflammation, oncogenesis, cytokines, adults
Abstract
Previous researchers have recorded that inflammation plays a critical role in tumorigenesis and some of the underlying molecular mechanisms have been elucidated. It is estimated that under-lying infections and inflammatory reactions are associated with a rate of 25% of all cancer cases. The role of inflammation in tumorigenesis is acceptable even in case where a direct etiological relationship with inflammation has not yet been established since inflammatory micro-environment is an essential component of all tumors. Only a minority of all cancers are caused by mutations of progenitor series, while the vast majority (90%) is associated with physical mutations and environmental factors. Many environmental causes and risk factors of cancer are associated with some type of chronic inflammation. It has been reported that 20% of cancers is associated with chronic inflammations, 30% could be attributed to cigarette smoking and inhaled pollutants such as silica and asbestos and 35% in nutritional factors, of which 20% is associated with obesity. Although inflammation, increases the risk for cancer, it was found recently that in addition to necessity of a tumor initiator, tobacco consists a promoter due to its ability to cause chronic inflammation. Most solid malignancies occur in the elderly where old age and cellular aging have been found to be tumor promoters that act through inflammatory mechanisms. DNA damage accumulation and cell aging can strengthen chronic inflammation that promotes tumor genesis. Cancer-associated inflammation contributes to tumor growth via several mechanisms, including the induction of gene instability, changes in epigenetic events and subsequent improper gene expression, enhance cell proliferation and resistance to apoptosis of the initial cells, immune suppression, induction of tumor angiogenesis and tissue re-modeling, and finally metastasis.
Inflammation Types and Mechanisms of Carcinogenesis
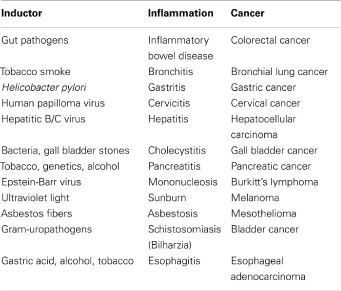
Various types of inflammation that vary depending on the cause, mechanism, effect and intensity can promote cancer growth. There have been found proven links between inflammatory process stages and cancer, such as inflammatory bowel and Crohn's disease and particularly ulcerative colitis, and colon cancer [1], persistent H. pylori infection and gastric cancer/MALT lymphoma, HBV and HCV infection and hepatocellular carcinoma, infection caused by Schistosoma and bladder cancer, Bacteroids infection and colon cancer [2,3], chronic reflux esophagitis in Barrett's esophagus and esophageal carcinoma [1], cervix infection caused by HPV and cervical cancer, prostatitis and prostate cancer, pancreatitis, and pancreatic cancer [4].
Inflammatory response triggered by infection preceding the development of the tumor and is a part of the host’s natural defense, and its target is pathogens elimination. However, pathogenic oncogenetic agents prevent the host’s immunity, and create persistent infections that associated with low-grade but chronic inflammation. In contrast, acute inflammation induced by specific microbial species was used by Coley with some success in treating cancer in1890 and has recently applied for bladder cancer treatment. This phenomenon that makes bladder cancer uniquely sensitive to acute inflammation, even when was promoted by chronic inflammation, is currently unknown [5].
Another type of chronic inflammation that precedes cancer development caused by immune de-regulation and autoimmunity. An example is inflammatory bowel disease which increases to a large degree colon cancer risk [6]. However, all inflammatory diseases do not increase cancer risk and some of them, such as psoriasis, may reduce such a risk [7].
Chronic inflammation may be caused by environmental exposure to tobacco material and other irritants that can lead to Chronic Obstructive Pulmonary Disease (COPD) that is associated with a higher risk of lung cancer development [8]. Inhaled asbestos particles or silica, can lead to lung cancer but without any obvious mutagenic action. Such particles, however, can trigger an inflammatory response through effects in pre-interleukin-1b induced by inflammation-particle that consists of caspase 1 PYCARD gene (N-terminal region PYRIN-PAAD-DAPIN and C-terminal caspase recruitment area), NALP (NOD similar subcontainer) and caspase 5, and this may mediate an oncogenic activity [9] (Figure 1).
Figure 1. Inflammation types and carcinogenesis
A completely different inflammation type is the one who follows tumor development. Almost all solid malignant tumors trigger an internal inflammatory reaction that creates a pre-oncogenetic microenvironment [10]. Based on the continuous cell renewal and proliferation that are induced by inflammation-associated tumor, the tumors have been reported as ‘wounds that cannot heal’ [11]. That inflammation type is a reversible wound healing response and tissue regeneration. Furthermore dominant oncogenes such as V-Src or K-ras are incapable to induce cancer in adult mice unless they are accompanied by trauma and subsequent tissue regeneration [12].
Immune Cell and Tumorigenesis
As a result of different inflammation types, tumor microenvironment contains native immune cells such as macrophages, polymorphonuclear leukocytes (PMN), mast cells, bone marrow suppressor cells, dendritic and natural killer cells (NK), adaptive immune cells such as B-and T-lymphocytes, cancer cells and its surrounding layer that consists of fibroblasts, endothelial cells, pericytes and mesenchymal cells [13]. Those cells show interactions each other through a direct contact or through cytokines and chemokines production and act in autocrine or/and paracrine ways to control and configuration of the tumor growth. Expression of various immunological mediators and regulators as well as the abundance and state of various cell types activation in tumor microenvironment determine the direction of the balance and if an inflammation will promote tumor or an antitumor immunity will occur [14,15].
In established tumors this balance is inclined to inflammation that will promote tumor growth. However, it is difficult to estimate the overall impact of immunity and inflammation in the early oncogenetic stages, because in vivo models for examination of such phenomena regarding the initial tumor development are not available. Moreover, recent knowledge is based on tumor load measurement to a point where malignant cells have already been escaped in an earlier stage by surveillance mechanisms. Malignancy is often associated with suppression of an effective T-cell response and an enhanced humoral immunological response. Undifferentiated tumor nature and the ability of cancer cells to evade the immune cells and suppress directly host’s immunity, possibly contribute to the failure of a reaction-response against the tumor [15].
Despite the presence of autologous T-effector cells, those cells are suppressed by an expanding population of regulatory T-cells (T regs) that are accumulated in tumor sites. Those regulatory T-cells can be activated or engaged by dendritic cells that are present in the tumor [16]. Another cell population that appears to play a role in suppressing of lymphocytes that infiltrate tumors (TILs) is myeloid suppressor cells that derived from bone marrow (MDSCs). Those cells initially, were recognized for their suppressive capacity, however it is suggested that participate in pro-inflammatory processes and are also capable of promoting angiogenesis [17].
It has been found that tumor-related inflammation characterized by pro-inflammatory and anti-inflammatory signals that allow tumors to grow and to escape from immune surveillance [18]. In conclusion, tumor evolution/growth, inflammation and immunity against the tumor coexist in different parts of its development, and these environmental and micro-environmental conditions determine the balance between them [19,20].
Leukocytes’ infiltration that varies in composition and distribution is present in the majority of tumors and is implicated in tumorigenesis, tumor growth, invasion and metastasis [10]. However, genetic experiments in mice have shown that inflammatory reaction that is supported by innate immune cells is crucial for adaptive immune response activation and also that is capable of eliminating tumor’s growth. It has been suggested that immune cells continually recognize and destroy 'nascent' cancer cells but due to genetic instability, new cancer cells avoid the immunosurveillance mechanisms [21]. In this context, several studies have focused on discovery of the mechanisms that lead to immune-escape and have given emphasis on underlying inflammation that is associated with established tumors and which is mainly oriented to the coordination of adaptive immunological response [22].
The most common cells that are present in tumor microenvironment, are macrophages associated with tumor (TAMs) and T-cells. TAMs cells, are the most important factors of inflammation and cancer, and consist a major source of cytokines [10]. Macrophages are distinguished in M1 and M2 types [23]. M1 type is activated by interferon gamma (IFN-γ) and microbial products, can express high levels of pro-inflammatory cytokines, such as TNF-α, IL-1, IL-6, IL-12, IL-23, molecules of the Major Histocompatibility System (MHC), inducible nitric oxide synthase (i-NOS), and are capable of killing pathogens and contribute to anti-tumor immune responses. In contrary to the mentioned activities of M1 cells, M2 type, that induced in vitro by cytokines IL-4, IL-10 and IL-13 down-regulates MHC class II and IL-12 expression, and shows an increased expression of anti-inflammatory cytokine IL-10, receptor scavenger A and arginase. Most TAMs assumed that have a M2 phenotype as they promote tumor angiogenesis, and tissue remodeling [23]. However, the most tumor promoting cytokines are M1-cytokines, while IL-10 that is a M2-cytokine, may be act as a tumor suppressor, as it was found to be present in colorectal cancer [14].
TAMs particularly promote tumor growth and may be are required for angiogenesis, invasion and metastasis [24]. T-cells are characterized by tumor suppressor and tumor promoting action [15]. Impairment of T-cells, or disruption of specific cytotoxic mechanisms can lead mice to be render more susceptible to spontaneous or chemical carcinogenesis [25]. However, it has been shown that many subsets of T cells that are present in solid tumors are involved in tumor’s promotion, progression or metastasis, including CD8 + T cells [26], Th1-cells that produce INF-γ [27], Th2-cells [28] and Th17-cells [9]. NK cells are the only cell subtype that do not show lacking pre-tumorigenic role. Similar to TAMs, tumorigenic functions of T cells are mediated by cytokines, whereas cytokines and cytotoxic mechanisms mediate T cells anti-tumorigenic functions [25].
Other immune cells also influence tumorigenesis. PMN act as tumor promoters and suppressors, depending on their differentiation status and the presence of transforming growth beta- factor (TGF-b) [29]. B-and mast cells are important contributors to the immuno-mediated tumor growth and conventional macrophages and dendritic cells are important for antigen presentation and T-cells activation during immune against tumors and for producing cytokine and induction of immunosuppression in established tumors. Different cytokines can either promote or inhibit tumor development and progression, regardless of their source [30].
Through activation of various downstream effector, such as transcription nuclear factor NF-kB, protein activator-1 (AP-1), STAT, SMAD factors and caspases, cytokines are able to control the immune and inflammatory environment either through anti-tumor immunity (IFN-c, TRAIL, IL-12) or enhance tumor development (IL-6, IL-17, IL-23) and also have direct effects on tumor cells growth and survival (TRAIL, IL-6, FasL, TNF-α, EGFR ligands, TGF-b) [14]. In conclusion, chronic inflammation is recognized as a pathological basis for the development of most tumors including those that are not causally related with infections and an underlying inflammation has recently been proposed as the seventh feature of cancer [31] (Figure 2).
Figure 2. Immune cells and tumorigenesis
Inflammation and Tumor Initiation
Inflammation mechanism involves innate and adaptive immunity that is characterized by the co-ordinated transfer through blood cells and soluble mediators to the injured tissues. After the elimination of pathogen and wound, the inflammation subsides. However, in case of an unsolved inflammation due to any failure in precise control of the immune response, inflammation may continue to disrupt the cellular microenvironment, leading to changes in genes that are associated with cancer and post-translational modifications in cell cycle’s signaling key-proteins, DNA repair and apoptosis. Mononuclear inflammatory cells are often present in a very early stage of tumor development in close relation with hyperplasia and atypia areas [32,33].
These findings support the assumption that mononuclear inflammatory cells themselves are the principle force that contributes to tumor initiation or/and in tumor initial development. In addition to macrophages, mast cells and PMN may also support tumor development resulting in the up-regulation of non-specific pro-inflammatory cytokines such as INF-γ, TNF-α, IL-1a, IL-1b and IL-6 [34,35]. Similarly, activated nuclear transcription factor NF-kB is one of the main ligands between inflammation and tumorigenesis and maybe the key that allows pre-neoplasmatic and malignant cells to escape from apoptosis. Consequently, all those factors can act as carcinogenesis primers and promoters due to proliferation of epithelial cells [2,36]. Tumor initiation consists a process in which normal cells acquire the first mutation hit that send them to tumorigenesis path of providing benefits for increasing and survival benefits against surrounding cells. However, in most cases, a single mutation is insufficient and many cancers require at least 4 or 5 mutations. The oncogenic mutations may occur in differentiated epithelial cells, such as hepatocytes that are able to proliferate and have a long life to allow further ‘hits’ mutations. It has been suggested that an inflammatory microenvironment can increase the mutation rates, in addition to strengthening the proliferation of mutant cells [37,38].
All inflammatory cells such as PMN, monocytes, macrophages, eosinophils, dendritic cells, mast cells and lymphocytes are recruited after injury or infection and may contribute to the onset and progression of cancer. Molecules-keys that link inflammatory response with genetic variations are prostaglandins (PGs), cytokines, transcription factor NF-kB, chemokines and angiogenic factors. Chronic inflammation is often accompanied by an increased production of reactive oxygen species (ROS) in tissue [39], which may change the signal transduction cascades and are able to induce changes in transcription factors, such as NF-kB, NF-E2/rf2 or Nrg2 (nuclear factor erythroid 2/related factor 2) and AP1 factor, which direct is involved in cellular stress responses [40,41].
2021 Copyright OAT. All rights reserv
The main chemical effectors, ROS, and reactive nitrogen intermediates (RNI), may act directly or indirectly, through reactions with other chemical or structural cells components and their derivatives can also activate factors such as NF-kB factor, resulting in production of other pro-inflammatory cytokines which in turn enhance inflammation and thus more ROS production and can attract other inflammatory cells leading to a secondary enhancing of lesion. Main ROS sources in all cells are mitochondria, Cyt P450 and peroxysomes [42]. Under normal circumstances exists an endogenous stabilize production of ROS and RNI that interact as 'signaling molecules' for cell metabolism, cell cycle and intercellular communication pathways [43]. That balance between the beneficial and/or adverse effects of ROS is a critical event in living organisms. Actually, redox homeostasis is, in vivo, the main protective process against cell death. For controlling the balance between production and removal of ROS and RNI exists a range of protective molecules and systems that are known as ‘anti-oxidant defenses’. Those enzymes such as super-oxide dismutase, catalase, glutathione peroxidase and glutathione-S-transferase are proteins that neutralize transition metals, glutathione, cysteine, thioredoxin, etc. [44]. Oxidative stress occurs in cases where ROS and RNI production in a system exceeds the system's ability to neutralize and eliminate them [45].
Under oxidative stress conditions, ROS and RNI act as toxic substances that can react with proteins, carbohydrates and lipids with consequent alteration in intracellular and intercellular homeostasis, leading to possible cell death and regeneration. The cells could respond to these ‘attack’ by enhancing their antioxidant capacity or via activation of the caspase system which induces programmed cell death (apoptosis). In cases where those protective systems fail exists a probability to have a mutant as a consequence of high disturbance in redox balance that is evolving through ROS and RNI in cell nucleus. Initiation of carcinogenesis that mediated by ROS and RNI after chronic inflammation may be direct, i.e. by oxidation, nitration, halogenation of the nuclear DNA, RNA and lipids, or may be mediated by ROS and RNI products as well as proteins, lipids and carbohydrates that can form complexes with DNA. ROS may also increase the expression of transcription factors, such as c-fos, c-jun and oncogenes that involved in neoplastic transformation [42].
Activated inflammatory cells act as a source of ROS and RNI and are capable of inducing DNA damage and genomic instability. ROS pre-neoplastic action is associated with their ability to induce DNA damage [46] that can lead to interruption or induction of transcription, induction of signal transduction pathways, transcription errors and genomic instability, procedures related to carcinogenesis, and also through proto-oncogenes activation, tumor suppressor genes inactivation, while in this procedure participate the oxidative damage of mitochondria [46,47]. Mitochondrial DNA fragments have been found in nuclear DNA suggesting a potential oncogene activation mechanism. Damage to mitochondrial DNA causes mitochondrial respiratory dysfunction which in turn increases the production of hydroxyl-radicals that is the major source of DNA oxidative damage [48]. Mutations and altered expression in mitochondrial genes that encode for I, III, IV and V complexes and in mitochondrial DNA hypervariable regions have been found in various human cancers [49].
However, it is not clear whether ROS and RNI that are produced and released from PMN or macrophages, especially during an acute inflammatory response, are sufficiently long-lived to diffuse through the extracellular matrix, enter epithelial cells and react with the packaged DNA, into chromatin [50]. Alternatively, inflammatory cells may use cytokines such as TNF-α to induce ROS accumulation in neighboring epithelial cells. Inflammatory cells produce cytokines and chemokines that promote tumor cells generation, in addition to producing an autocrine growth factor by the tumor cells themselves. ROS and RNI are produced under the influence of pro-inflammatory cytokines in phagocytic and non-phagocytic cells by activating of protein kinase signaling. Thus TNF-α increases ROS formation by PMN and other cells, whereas IL-1-b, TNF-α and IFN-γ induce iNOS expression in inflammatory and epithelial cells. IL-8, another inflammatory chemokine derived that from monocytes, macrophages and endothelial cells is involved in colorectal, bladder, lung and stomach cancer progression and metastasis [50].
Chronic inflammation is closely associated with angiogenesis, whereas macrophages, fibroblasts, platelets and own tumor cells are the principal source of angiogenic factors such as basic fibroblast growth factor (b-FGF), endothelial growth factor (VEGF ) and PGE-1 and E-2, in addition to inflammatory cytokines, chemokines and nitric oxide (NO) [51].
A significant interaction and synergy between those mediators of inflammation leads to cancer development. PGs that are derived from arachidonic acid oxidative metabolism in inflammatory cells, contribute to cancer development, as are able to induce the expression of inflammatory cytokines which in turn increase ROS and RNI production [52].
Cyclooxygenase-2 (COX-2) consists a main enzyme that responses to inflammation through PGs synthesis in monocytes and macrophages, but is also expressed in non-inflammatory cells such as fibroblasts, epithelial and endothelial cells. It consists an inducible enzyme and its expression is regulated by NF-kB factor that mediates tumorigenesis. COX-2 is involved in cancer biology and is upregulated in many types of cancer such as colorectal, breast, lung, pancreas, esophagus, head and neck [53-55], lower and high-level grade astrocytomas [56], melanomas [34], and plays an important role in lung adenocarcinoma development [57]. Studies have recorded a 40-50% reduction rate in mortality from colorectal cancer after taking NSAIDs compared to patients who did not receive them. This has been attributed to their ability to inhibit COX-2 that converts arachidonic acid into PGs [53].
Much research has been done whether immune-mediated mechanisms, unlike dietary and environmental mutagens are the critical main forces regarding tumor initiation [58]. However, p53 mutations, possibly caused by oxidative damage, and were found in cancer cells and in inflammatory non-dysplastic epithelium in cancer-associated colitis, suggesting that chronic inflammation causes changes in the genome [59].
Mutagenesis induced by inflammation can lead to inactivation or suppression of Mismatch Repair Genes (MMR) of DNA, whereas ROS can also cause direct oxidative inactivation of MMR enzymes [58,60]. In case MMR genes system has been deregulated, mutagenesis induced by inflammation is enhanced and also several tumor suppressor genes such Tgfbr 2 and Bax that have microsatellite sequences is possible to be inactivated [60].
Another mechanism that associates inflammation with oncogenic mutations is the upregulation of cytidine deaminase (AID) activation, an enzyme that promotes the rotation rate of immunoglobulin gene by catalyzing the deamination of cytosines in DNA. In B-cells AID is overexpressed in many cancers of various origins and its expression is induced by inflammatory cytokines in a NF-kB-dependent pathway or via TGF-b [61]. AID induces genetic instability and increases the probability of mutation acting in double-stranded DNA, introducing mutations at critical cancer genes, including Tp53, c-Myc and Bcl-6 [60], and contributes to the development of lymphomas, gastric, and liver cancers [61,62].
Other mechanisms of mutagenesis that are induced by inflammation have been proposed, and include the effects of inflammation in non-homologous recombination and through NF-kB-mediated inactivation of p53 dependent surveillance genome [60]. In those mechanisms by which inflammation may enhance tumor initiation are included the production of growth factors and cytokines that can give a like-stem cell phenotype in tumor progenitor cells, or to stimulate stem cells expansion which leads to cell space widening that is targeted by environmental mutagens. Indeed, STAT3 is associated with both the reprogramming of stem cells and their renewal [63], while NF-kB factor can enhance Wnt/b-catenin signaling in colon crypts [64]. TNF-α pro-inflammatory cytokine promotes b-catenin nuclear entry during inflammation that associated with gastric cancer in the absence of any mutations in Wnt/b-catenin components pathway [65].
The link between inflammation and cancer initiation is ‘no one way’ and exists also evidence that DNA damage can lead to inflammation and thus is able to promote tumorigenesis. An example is the model of hepatocellular cancer that induced by carcinogenic diethyl nitrosamine (DEN), wherein DNA damage contributes to necrotic cell death that leads to an inflammatory reaction which promotes tumor’s the growth [66] (Figure 3).
Inflammation and Tumor’s Promotion
This process is characterized by tumor growth that is initiated by a single original cell to a fully expanded primary tumor. Initial tumor growth depends on an increased cell proliferation and a reduced cell death. Both processes are stimulated by inflammation mechanisms. In fact many of the reinforcing effects of inflammation on cancer are obvious at the tumor promotion level and many known tumor promoters are important inducers of inflammation [2].
Tumor’s promotion that induced by inflammation mechanisms may occur early or late during tumor development, and inflammation can lead to precancerous lesions activation that were inactive for many years. The mechanisms by which inflammation affects tumor’s promotion are innumerable and besides increased proliferation and survival may also include an ‘angiogenic switch’ that allows a small latent tumor having blood supply that is necessary for the subsequent growth stage [67].
Cytokine Signaling Pathways and Targets-Genes in Tumor Promotion
Production of cytokines that promote tumors by immune/inflammatory cells that activate transcription factors such as NF-kB, STAT 3 and AP-1 in premalignant cells for genes induction that stimulate cell proliferation and survival, is an important mechanism of tumor’s promotion. Initial data for tumor-promoting inflammation have been derived from experimental animal studies. Although a contradictory fact in time, TNF-α factor was required for two-stage carcinogenesis on skin [68]. TNF-α factor activates transcription factors AP-1 and NF-kB, but on skin its tumor promoting effects are mediated by AP-1factor [69], which was identified as a transcription factor whose activity is stimulated by TRA (tetradeca-null-phorbol-acetate) that is a classical tumor promoter [70]. Instead, transcription factor NF-kB inhibits skin cancer development [71]. Thus, despite the fact a given cytokine can activate several transcription factors, its activity in tumor promotion may be mediated by only one of them and is antagonized by someone else. Among various transcription factors that are part of this mechanism, NF-kB and STAT 3 factors are activated in the majority of cancers and act as non-classical oncogenes, whose activation in malignant cells is rarely the result of direct mutations and instead it depends on signals that are generated by neighboring cells, or more rarely by activating mutations of upstream signaling components. NF-kB and STAT 3 factors activate genes that control cell survival, proliferation, growth, angiogenesis, invasiveness, mobility and chemokines and cytokines production [72,73].
It has been found in experimental animal studies that various cytokines such as IL-6, IL-11, hepatocyte growth factor (HGF), EGF and tyrosine kinases such as c-Met, Src, promote tumori-genesis [74]. Another cytokine-tumor promoter is IL-23 [75] that is expressed primarily by TAMs and its expression depends on STAT 3 and NF-kB factors [76]. Most of the genes that mediate tumor-promoting functions and especially the functions of NF-kB, STAT 3 and AP-1 factors have not been fully determined and most possible pro-tumor-promoting effects of those transcription factors are affected by multiple effectors. Some targets may be controlled by multiple transcription factors and may be more important in one cell type than in another. Expression of antiapoptotic proteins, for example, Bcl-2 and Bcl-XL is promoted by NF-kB and STAT 3 factors and c-IAP1, c-IAP 2, Mcl-1, c-FLIP factors and survivin expression as well [2,77]. NF-kB and STAT 3 factors interfere with p53 gene synthesis and attenuate p53, mediated-genomic surveillance, representing another possible tumor-promoting mechanism [60]. STAT 3 factor controls Cyclin D1, D2, B and c-Myc proto-oncogenes expression and through those can induce cell proliferation [77,78]. Although Cyclin D and c-Myc is also considered to be regulated by NF-kB factor, inactivation of IKKb inhibitor in enteric cells is not involved in cellular proliferation [79] and in Ras-transformed keratinocytes [71] or DEN (diethyl-nitro-za-min)-activated hepatocytes, while NF-kB inhibition actually enhances Cyclin D expression and cell proliferation [80].
Oncogenes are involved in cell growth regulation. Among their pleiotropic effects are included a precancerous microenvironment induction through a persistent promotion of an inflammatory environment [81,82]. Chemokines and cytokines that promote tumorigenesis act by autocrine and paracrine pathways for inflammatory cells recruitment in tumor’s microenvironment. Chronic inflammation’s perpetuation is achieved mainly by means of positive feedback brackets which concern inflammatory cells that produce cytokines and induce chemokines synthesis in stromal and malignant cells leading to prolonged inflammatory cells recruitment in tumor micro-environment. In this respect, TAMs, MDSCs, T regs and Th17-cells are the major immunological cell subtypes. Recruitment of myeloid cells is regulated by multiple pathways including CCL2 - CCR2, CCL1 - CXCR2, S 100A-protein-RAGE and IL-1-IL-1R, interactions. CCR6 signaling is critical for infiltration with Th 17-cells, whereas T regs-cells are recruited mainly by CCR4 and CCR7 chemokines [83]. In some cases those critical chemokines are not produced by cancer cells but are induced in cancer-associated fibroblasts (CAFs) based on interaction with tumor cells [84] (Figure 4).
Figure 3. Tumor initiation
Figure 4. Tumor promotion
References
- van der Woude CJ, Kleibeuker JH, Jansen PL, Moshage H (2004) Chronic inflammation, apoptosis and (pre-) malignant lesions in the gastro-intestinal tract. Apoptosis 9: 123-130. [Crossref]
- Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441: 431-436. [Crossref]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, et al. (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15: 1016-1022. [Crossref]
- Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, et al. (2000) Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 60: 3333-3337. [Crossref]
- Rakoff-Nahoum S, Medzhitov R (2009) Toll-like receptors and cancer. Nat Rev Cancer 9: 57-63. [Crossref]
- Waldner MJ, Neurath MF (2009) Colitis-associated cancer: the role of T cells in tumor development. Semin Immunopathol 31: 249-256. [Crossref]
- Nickoloff BJ, Ben-Neriah Y, Pikarsky E (2005) Inflammation and cancer: is the link as simple as we think? J Invest Dermatol 124: x-xiv. [Crossref]
- Punturieri A, Szabo E, Croxton TL, Shapiro SD, Dubinett SM (2009) Lung cancer and chronic obstructive pulmonary disease: needs and opportunities for integrated research. J Natl Cancer Inst 101: 554-559. [Crossref]
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, et al. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674-677. [Crossref]
- Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454: 436-444. [Crossref]
- Dvorak HF (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315: 1650-1659. [Crossref]
- Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, et al. (2007) Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291-302. [Crossref]
- de Visser KE, Eichten A, Coussens LM (2006) Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6: 24-37. [Crossref]
- Lin WW, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117: 1175-1183. [Crossref]
- Smyth MJ, Dunn GP, Schreiber RD (2006) Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol 90: 1-50. [Crossref]
- Bacchetta R, Gambineri E, Roncarolo MG (2007) Role of regulatory T cells and FOXP3 in human diseases. J Allergy Clin Immunol 120: 227-235. [Crossref]
- Talmadge JE, Donkor M, Scholar E (2007) Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev 26: 373-400. [Crossref]
- Murdoch C, Muthana M, Coffelt SB, Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8: 618-631. [Crossref]
- Bui JD, Schreiber RD (2007) Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr Opin Immunol 19: 203-208. [Crossref]
- Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, et al. (2008) Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A 105: 652-656. [Crossref]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3: 991-998. [Crossref]
- Mantovani A, Sica A, Allavena P, Garlanda C, Locati M (2009) Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol 70: 325-330. [Crossref]
- Sica A, Allavena P, Mantovani A (2008) Cancer related inflammation: the macrophage connection. Cancer Lett 267: 204-215. [Crossref]
- Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124: 263-266. [Crossref]
- Swann JB, Smyth MJ (2007) Immune surveillance of tumors. J Clin Invest 117: 1137-1146. [Crossref]
- Roberts SJ, Ng BY, Filler RB, Lewis J, Glusac EJ, et al. (2007) Characterizing tumor-promoting T cells in chemically induced cutaneous carcinogenesis. Proc Natl Acad Sci U S A 104: 6770-6775. [Crossref]
- Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, et al. (2006) IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med 203: 1391-1397. [Crossref]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, et al. (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16: 91-102. [Crossref]
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, et al. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell 16: 183-194. [Crossref]
- Andreu P, Johansson M, Affara NI, Pucci F, Tan T, et al. (2010) FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 17: 121-134. [Crossref]
- Mantovani A (2009) Cancer: Inflaming metastasis. Nature 457: 36-37. [Crossref]
- Mantovani A, Schioppa T, Porta C, Allavena P, Sica A (2006) Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev 25: 315-322. [Crossref]
- Pollard JW (2004) Tumour-educated macrophages promote tumor progression and metastasis. Nat Rev Cancer 4: 71-8.
- Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G (2006) Inflammation and cancer: how hot is the link? Biochem Pharmacol 72: 1605-1621. [Crossref]
- Robinson SC, Coussens LM (2005) Soluble mediators of inflammation during tumor development. Adv Cancer Res 93: 159-187. [Crossref]
- Naugler WE, Karin M (2008) NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev 18: 19-26. [Crossref]
- Tokuhata GK, Lilienfeld AM (1963) Familial aggregation of lung cancer in humans. J Natl Cancer Inst 30: 289-312. [Crossref]
- Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759-767. [Crossref]
- Babior BM (2000) Phagocytes and oxidative stress. Am J Med 109: 33-44. [Crossref]
- Closa D, Folch-Puy E (2004) Oxygen free radicals and the systemic inflammatory response. IUBMB Life 56: 185-191. [Crossref]
- Kwak MK, Kensler TW (2010) Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol 244: 66-76. [Crossref]
- Marnett LJ, Riggins JN, West JD (2003) Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest 111: 583-593. [Crossref]
- Nathan C (2003) Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest 111: 769-778. [Crossref]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, et al. (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44-84. [Crossref]
- Sies H, Cadenas E (1985) Oxidative stress: damage to intact cells and organs. Philos Trans R Soc Lond B Biol Sci 311: 617-631. [Crossref]
- Marnett LJ (2000) Oxyradicals and DNA damage. Carcinogenesis 21: 361-370. [Crossref]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160: 1-40. [Crossref]
- Berlett BS, Stadtman ER (1997) Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 272: 20313-20316. [Crossref]
- Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C (2007) Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer 121: 2381-2386. [Crossref]
- Zouki C, József L, Ouellet S, Paquette Y, Filep JG (2001) Peroxynitrite mediates cytokine-induced IL-8 gene expression and production by human leukocytes. J Leukoc Biol 69: 815-824. [Crossref]
- Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD (1997) The codependence of angiogenesis and chronic inflammation. FASEB J 11: 457-465. [Crossref]
- Prescott SM, Fitzpatrick FA (2000) Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta 1470: M69-M78. [Crossref]
- Lin DT, Subbaramaiah K, Shah JP, Dannenberg AJ, Boyle JO (2002) Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck 24: 792-799. [Crossref]
- Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, et al. (1998) Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 93: 705-716. [Crossref]
- Half E, Tang XM, Gwyn K, Sahin A, Wathen K, et al. (2002) Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res 62: 1676-1681. [Crossref]
- Shono T, Tofilon PJ, Bruner JM, Owolabi O, Lang FF (2001) Cyclooxygenase-2 expression in human gliomas: prognostic significance and molecular correlations. Cancer Res 61: 4375-4381. [Crossref]
- Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, et al. (1998) Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res 58: 4997-5001. [Crossref]
- Hussain SP, Hofseth LJ, Harris CC (2003) Radical causes of cancer. Nat Rev Cancer 3: 276-285. [Crossref]
- Kraus S, Arber N (2009) Inflammation and colorectal cancer. Curr Opin Pharmacol 9: 405-410. [Crossref]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30: 1073-1081. [Crossref]
- Okazaki IM, Kotani A, Honjo T (2007) Role of AID in tumorigenesis. Adv Immunol 94: 245-273. [Crossref]
- Takai A, Toyoshima T, Uemura M, Kitawaki Y, Marusawa H, et al. (2009) A novel mouse model of hepatocarcinogenesis triggered by AID causing deleterious p53 mutations. Oncogene 28: 469-478. [Crossref]
- Chen X, Xu H, Yuan P, Fang F, Huss M, et al. (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106-1117. [Crossref]
- Umar S, Sarkar S, Wang Y, Singh P (2009) Functional cross-talk between beta-catenin and NFkappaB signaling pathways in colonic crypts of mice in response to progastrin. J Biol Chem 284: 22274-22284. [Crossref]
- Strid J, Roberts SJ, Filler RB, Lewis JM, Kwong BY, et al. (2008) Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol 9: 146-154. [Crossref]
- Oguma K, Oshima H, Aoki M, Uchio R, Naka K, et al. (2008) Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J 27: 1671-1681. [Crossref]
- Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res 66: 605-612. [Crossref]
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, et al. (1999) Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med 5: 828-831. [Crossref]
- Eferl R, Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859-868. [Crossref]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, et al. (1987) Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 49: 729-739. [Crossref]
- Zhang JY, Green CL, Tao S, Khavari PA (2004) NF-kappaB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev 18: 17-22. [Crossref]
- Grivennikov SI, Karin M (2010) Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 21: 11-19. [Crossref]
- Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9: 798-809. [Crossref]
- Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140: 883-899. [Crossref]
- Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, et al. (2009) Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15: 114-123. [Crossref]
- Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, et al. (2006) IL-23 promotes tumour incidence and growth. Nature 442: 461-465. [Crossref]
- Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7: 41-51. [Crossref]
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, et al. (2009) gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15: 91-102. [Crossref]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, et al. (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285-296. [Crossref]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M (2005) IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121: 977-990. [Crossref]
- Croce CM (2008) Oncogenes and cancer. N Engl J Med 358: 502-511. [Crossref]
- Grivennikov SI, Karin M (2010) Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev 20: 65-71. [Crossref]
- Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, et al. (2009) Chemokines and chemokine receptors: an overview. Front Biosci (Landmark Ed) 14: 540-551. [Crossref]
- Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA (2009) Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One 4: e7965. [Crossref]