The coronavirus 2 (SARS-CoV-2) has resulted in an international pandemic. The SARS-CoV-2 affects cardiovascular, digestive and urogenital systems. In an attempt to develop a multimodal and targeted approach to the pathophysiology associated with viral lung injury, we reviewed lung histopathology, inflammation, surfactant biology and pathophysiology related to viral associated acute lung injury (ALI / ARDS). Histopathology of viral pneumonia/ARDS cases of the past 100 years has revealed that lung parenchymal and vascular pathologic changes described in the 1918 influenza pandemic, are no different from the histopathology observed in other viral pandemics. Given the inflammatory storm which can occur in COVID-19 infection, the patient is a candidate to develop the classic multi-organ dysfunction and / or failure (MOD/F) which may well include ALI and ARDS. Because there is a well described temporal change in the pathophysiology associated with ALI /ARDS, a “one size fit al” remedy will not suffice, hence our attempt at a targeted functional approach. For instance, variable results in adults treated with surfactant have relegated the use of surfactant in adult ARDS to an uncertainty. We speculate that early and repeated surfactant installation in adults is required in adults. Surfactant may also have beneficial effects on the inflammatory process in the ARDS lung. The increased clotting tendency associated with the inflammation (and ARDS), particularly the effect on the lung vasculature (acute pulmonary hypertension and increased dead space), causes mechanical pressure overload (and failure) of the right ventricle and mechanical respiratory failure. Active management of these should include inhibition of the accelerated coagulation and thrombolysis (via nebulization) and early inotropic support.
COVID-19, angiotensin-converting enzyme 2 receptor, surfactant, ARDS/ALI, anticoagulant agents
A recently identified human pathogen, acute respiratory syndrome coronavirus 2 (SARS-CoV-2 or COVID-19), a beta coronavirus closely related to acute respiratory syndrome coronavirus (SARS-CoV), has resulted in a pandemic with excessive mortality [1]. The SARS-CoV-2 is highly contagious and uses the angiotensin-converting enzyme 2 (ACE2) receptor for host cell entry with the aid of its surface spike (S) protein. The COVID-19 S protein binds ACE2 with higher affinity than does SARS-CoV [1]. In the respiratory system, ACE2 receptors are mainly expressed by type II alveolar cells (AT2) but are also found in the oral cavity. However, because ACE2 receptors are also expressed in various other organs, including the cardiovascular, digestive and urogenital systems, it inter alia explains the widespread organ pathology associated with SARS-CoV-2 [2,3].
It is claimed that the virus induces an atypical lung injury, and that symptoms, signs and functional tests cannot not be explained by impaired pulmonary parenchymal dysfunction alone. Furthermore, “recent” observations suggest that respiratory failure in COVID-19 is driven by and neither similar to the acute respiratory distress syndrome (ARDS), but that microvascular thrombotic processes play a unique and central role in its pathophysiology [4-6].
We briefly review lung histopathology, inflammation and surfactant biology related to past-, present viral-, including coronavirus induced acute lung injury or acute respiratory distress syndrome (ALI/ARDS). Furthermore, like Nieman et al, we are of the opinion that a physiological approach to the management of the pathophysiology is warranted [7]. This approach applies to the less severe ALI (either referring to a low Murray count), which may include single segment lung involvement [8] or mild ARDS (according to the PaO2/FiO2 ratio) [8] and the more severe lung involvement (higher Murray score of worse oxygenation ratio) [9,10]. We also examined the role for exogenous lung surfactant replacement therapy, with or without adjunctive appropriate anticoagulant agents and cardiovascular support in the treatment of the pathophysiological processes associated with virus induced lung pathology.
However, when reviewing the histology of viral pneumonia, be it direct viral infection or the result of interleukin induced inflammatory lung injury (acute lung injury (ALI) or acute respiratory distress syndrome, (ARDS)), i.e., the expression of lung injury associated with multi-organ inflammatory disease and failure (MOD/F)), publications are often not clear with reference to the stage of the disease process being addressed. Although the generic description of a direct viral pneumonia from a standard text reads as follows: “All causal agents produce essentially similar morphologic patterns. The pneumonic involvement may be quite patchy or may involve lobes bilaterally or unilaterally. The affected areas are red-blue, congested and sub crepitant. The pleura is smooth, and pleuritis and pleural effusions are infrequent”, there is a clear difference in the histology and pathophysiological processes depending on the time frame and course of the disease” [10,11].
The histology pattern is related to the clinical severity of the disease. The general predominant finding is that of an interstitial inflammatory reaction, virtually localized within the walls of the alveoli. The alveolar septa are widened and edematous and usually have mononuclear inflammatory infiltrates of lymphocytes, histiocytes and occasionally plasma cells. In acute cases, neutrophils may also be present. The alveoli may be free from exudates but in many patients, there is intra alveolar proteinaceous material, a cellular exudate and characteristically pink hyaline membranes lining the alveolar walls, - similar to those seen in hyaline membrane disease in the infant. These changes reflect alveolar damage similar to those seen in ARDS [12]. Thus, the above quoted section suggests that, unless the pathologist is informed of the complete course of the disease, the acute viral pneumonic process may well be confused with the general inflammatory driven ALI /ARDS.
In the discussion to follow, it was often difficult to distinguish the above permutations given the paucity of data regarding the time temporal course of the disease process. Hence this publication attempts to deal with the topic in a more generic fashion.
The worldwide outbreak of acute respiratory syndrome (SARS) caused by a new coronavirus (SARS-CoV), occurred between November 2002 and July 2003. Histopathologic examination of lung specimens from SARS-CoV victims revealed features of diffuse alveolar damage (DAD) with marked pulmonary oedema and hyaline membrane formation. Intra-alveolar organization and interstitial thickening with mild to moderate fibrosis were features of the later stage of the disease. Features of bronchiolitis obliterans-organizing pneumonia-like lesions were also noted in some patients [13].
As can be expected, different histological patters were noted when examining 8 cases [13]. Cases of shorter duration (10 or fewer days) demonstrated histological features of acute-phase DAD, airspace oedema, and bronchiolar fibrin, hyaline membranes, interstitial and intra-alveolar oedema and interstitial infiltrates of inflammatory cells. Vascular congestion occurred in 50% to 75% of the lung parenchyma of the acute phase DAD cases. Cases of longer duration (more than 10 days) demonstrated features of organizing phase DAD i.e. type II pneumocyte hyperplasia, squamous metaplasia, multinucleated cells and acute bronchopneumonia. However, the authors did not report on pulmonary blood vessel thrombi.
Hwang and associates described the pulmonary histologic features of SARS-CoV-positive cases [14]. Patients predominantly showed a DAD pattern of lung injury, with formation of hyaline membranes and interstitial thickening. Six cases predominantly showed an acute fibrinous and organizing pneumonia pattern of injury, as evidenced by formation of fibrin ‘balls’ within airspaces rather than hyaline membranes, and by an organizing pneumonia pattern with fibrosis. The remaining cases showed variable degrees of both patterns. Cases of shorter duration (14 days duration or less) had more widespread acute injury (exudative DAD) than those of longer duration (more than 14 days), while those of longer duration showed more extensive organization. Other associated histologic features, such as pneumocyte hyperplasia, squamous metaplasia, and multinucleated cells, were present in the SARS-CoV and comparison group. Injury of the pulmonary vasculature (intravascular fibrin thrombi and thrombo-emboli), were associated with lung parenchymal infarcts in many cases [14].
Publications on the histopathology of viral pneumonia/ARDS cases of the past 100 years revealed that the spectrum of pathologic changes, described in the 1918 influenza pandemic, was not significantly different from the histopathology observed in other viral pandemics, seasonal influenza, corona virus, including COVID-19 outbreaks (Table 1) [15-19]. Histopathology consistently revealed similar characteristic lung changes in viral-induced pneumonia/ARDS: Development of interstitial and alveolar oedema, hyaline membranes, focal necrosis of the alveolar wall and capillary thrombosis of the vessels in the alveolar wall and septa [15-19]. Alveolar epithelium undergoes necrotic changes and desquamation and vast numbers of desquamated cells are observed in the luminal spaces of alveoli, alveolar ducts and bronchioli. This, together with macrophages, containing phagocytosed cellular debris, is commonly seen. Intra-alveolar haemorrhage and oedema and near necrotic areas are characteristic features of virus pneumonia and are associated with the exudation of plasma and fibrin strands. With rare exceptions, the main lung histopathology conforms to what is known as DAD, i.e. the histological prototype of acute lung injury. The mechanism is believed to be endothelial and alveolar epithelial cell injury leading to fluid and cellular exudation, subsequent reparative fibroblastic proliferation and type II pneumocyte hyperplasia [16-18] (Figure 1) [19-23].
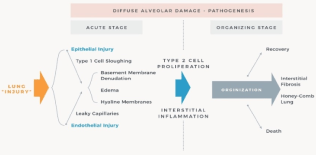
Figure 1. Schematic diagram of the pathogenesis of ALI/ARDS following lung injury. Figure was adapted from references [20-23]
Table 1. Summary of studies in viral-related ARDS and associated predominant lung histopathology of the past 100 years
First author (year) Reference |
Pandemic/Epidemic (Year) |
Number of patients with viral-related ALI/ARDS |
Timeline between onset and death |
Main histopathology findings |
LeCount (1919) [19] |
Influenza (1917/1918) |
1 case with pneumonia |
10 days |
Disseminated necrosis of the interalveolar microcapillaries and oedema in the lung tissue. Perhaps the cause of haemorrhages or haemorrhagic infarcts in the lung |
Wolbach (1923) [15] |
Influenza (1917/1918) |
Fatal cases with ‘virus’ lesions |
7-32 days |
Virus lesion at the start - desquamation and denudation of ECs of bronchi, bronchioles, AD and lining of alv, laying bare BM. Intense congestion of the blood vessels in these areas. Blood cells in the airways and alv with fibrin. HMs in alv spaces. Few leukocytes or phagocytes were found. HMs persisted and became organized with connective tissue
Acute inflammatory reaction in arterial walls in some cases with a fibrin deposit, necrosis of the walls of the vessel and cellular infiltration. Thrombosis was found in the blood vessels. Lymphatics were distended. Together with evidence of repair (fibrosis), acute bronchial inflammation persisted |
Louria (1959) [25] |
Influenza (1957-1958) |
N=4 with diffuse lung involvement |
NR |
Similar lung findings in above 4. Bloody fluid in trachea, bronchi and alveoli. Hyperemic alv capillaries. Parenchyma, dark red, congested, edematous and heavy. No thrombi.
Tracheitis, bronchitis and bronchiolitis with loss of normal ciliated epithelial cells and EC regeneration.
Alv spaces contained neutrophils and mononuclear cells, admixed with fibrin and oedema fluid. Acellular, HMs lined ADs and alv |
Franks (2003) [13] |
SARS-CoV (2002)
(Singapore) |
N=8 |
4 - 20 days
<10 days
>10 days |
Acute phase DAD; including HMs, interstitial and intra-alveolar oedema, interstitial infiltrates of inflammatory cells, and vascular congestion. Fibrin thrombi in small PA in 2/4 cases
Organizing phase DAD; characterized by interstitial and airspace fibroblast proliferation, accompanied by repair in the form of AT2 pneumocyte hyperplasia and squamous metaplasia. Fibrin thrombi in small PA in 3/5 cases |
Hwang (2005) [14] |
SARS (2003/Toronto) |
N=20 |
5 - 108 days
<14 days
>14 days |
Acute DAD
Fibrin thromboemboli 80%
Pulmonary infarcts 20%
Acute fibrinous and organizing pneumonia.
Fibrin thromboemboli 87%
Pulmonary infarcts 73% |
Nin (2012) [28] |
Influenza A
H1N1 (2009)
Multi-centre |
N=6 |
<10 days
>10 days (16-45 days) |
Exudative DAD (2/3); Proliferative DAD (1/3); alveolar haemorrhage (3/3)
Proliferative DAD (2/3); fibrosis (1/3); necrotizing bronchiolitis (1/3); alv haemorrhage (3/3)
Other findings included microthrombi or thrombi in large arteries |
Ng (2016) [30] |
MERS
(MERS-CoV) (2014) |
N=1 |
12 days |
Exudative DAD |
Menter (2020) [33] |
COVID-19
(SARS-CoV-2)
(Switzerland) |
N=21 |
0-16 days |
Exudative DAD with massive capillary congestion often accompanied by microthrombi despite anticoagulation. Superimposed bronchopneumonia (10/21); Pulmonary embolisms (4/21); Alveolar haemorrhage (3/21) |
Xu (2020) [3] |
COVID-19
(China) |
N=1 |
14 days |
Bilateral DAD with cellular fibro myxoid exudates
Right lung: desquamation of pneumocytes and HMs
Left lung: pulmonary oedema with HMs
Interstitial mononuclear inflammatory infiltrates, dominated by lymphocytes, in both lungs.
Atypical enlarged pneumocytes |
Carsana (2020) [35] |
COVID-19
(Northern Italy) |
N=38 |
5-31 days |
Exudative and proliferative phases of DAD; capillary congestion, necrosis of pneumocytes, HMs, interstitial oedema, pneumocyte hyperplasia and platelet-fibrin thrombi in small arterial vessels.
Inflammatory infiltrate: macrophages in alveolar lumens and lymphocytes in interstitium |
Ackermann (2020) [36] |
COVID-19 versus
Influenza A
Controls |
N=7 COVID-19
N=7 Influenza A
N=10 Controls |
< 10 days
< 21 days
< 6 days |
DAD: 100% COVID-19 & Influenza A
Vascular angiogenesis distinguished the lung pathobiology of Covid-19 from that of influenza virus
Lungs of influenza heavier
Both COVID-19 & influenza have vascular thrombi
Alveolar CAP microthrombi were 9 times as prevalent in patients with COVID-19
Similar ACE2 expression in alveolar and ET cells in COVID-19 and influenza |
ECs: epithelial cells; AD: alveolar duct; BM: basement membrane; HMs: hyaline membranes; NR: not recorded; DAD: diffuse alveolar damage; AT2: alveolar type 2 cell; PA: pulmonary arteries; ET: endothelial; CAP: capillary; MERS: Middle East respiratory syndrome coronavirus (MERS-CoV); SARS-associated coronavirus (SARS-CoV); COVID-19: coronavirus disease of 2019 (SARS-CoV-2).
Given the inflammatory storm [24] which can occur in COVID-19 infection, patients are candidates to develop the more classic multi-organ dysfunction and / or failure (MOD/F) which may well include ALI and ARDS [25].
The pathological changes to the lung associated with influenza viral pneumonia, have recently been reviewed [16]. The acute alveolar injury (DAD) caused by influenza virus infection is similar to that caused by many other agents which are noxious to alveoli. In the early stage, there is necrosis of alveolar epithelium, characterized by denudation of the alveolar septum and the presence of desquamated pneumocytes in the alveolar lumen. These desquamated cells are shrunken and show pyknosis or karyorrhexis and cytoplasmic vacuolation or hypereosinophilia. The alveolar lumina are flooded with oedema fluid with a variable mixture of fibrin and erythrocytes (intra-alveolar haemorrhage). In some alveolar lumina, there are many alveolar macrophages. Characteristically, alveoli and alveolar ducts are lined with hyaline membranes, consisting of fibrin-rich oedema fluid mixed with the cytoplasmic and lipid remnants of necrotic epithelial cells. The alveolar septa are widened due to hyperaemia of alveolar capillaries, interstitial oedema, and leukocyte infiltration, - mainly neutrophils but also eosinophils. These leukocytes also may be present in alveolar lumina. Fibrinous thrombi may be present in the capillaries of alveolar septa and alveolar ducts, as well as in small pulmonary blood vessels. Possibly, as a result of the thrombi, alveolar septa become necrotic. The late stage of influenza viral pneumonia is characterized by re-epithelization of the alveoli by type II pneumocytes (type II pneumocyte hyperplasia), interstitial fibrosis of alveolar septa, and infiltration by mononuclear leukocytes, predominantly lymphocytes and plasma cells. In addition to the above alveolar changes, the bronchioles show necrotizing bronchiolitis, characterized by epithelial necrosis, the formation of hyaline membranes, and infiltration by variable numbers of neutrophils. Changes to the trachea and bronchi are similar to those of uncomplicated influenza. Chronic changes of influenza pneumonia may include squamous metaplasia and interstitial fibrosis [16,19].
In 1923 Wolbach and Frothingham described autopsy findings of 26 cases of the 1917/18 influenza epidemic [15]. In what he labelled as ‘the virus lesion’, the early stages (of pneumonia) consisted in an injury to the epithelial cells of the smaller bronchi, the bronchioles, the alveolar ducts and the lining of the alveoli themselves, desquamation of this epithelium and denudation of the alveoli. They noted that “In some places considerable strips of desquamated epithelium were seen detached from the basement membrane and at the same time, there was intense congestion of the blood vessels in these areas, and although no actual injury to the blood vessels could be made out, there must have been an injury because the red blood cells had broken out into the lumen of the bronchial tree and the alveolar spaces and a slight amount of fibrin had been deposited. In addition, a hyaline-like membrane was found in the alveolar spaces”.
Interestingly, Wolbach and Frothingham also described the presence of air in the subcutaneous tissues of the neck, in some cases over the entire trunk, head and extremities. It was felt at the time that the acute interstitial emphysema resulted from the mechanical rupture of some of the alveoli which have become distended by acute alveolar emphysema. The air trapping could have been as a consequence of intra alveolar fibrin clots and hyaline membranes causing a ‘ball-valve’ effect which was aggravated by high generated negative intrathoracic pressure associated with spontaneous breathing efforts in patients who had low lungs compliance. Although not defined at the time, patient self-initiated lung injury (P-SILI) may have played a role in determining some of the structural lesions observed in these cases [15].
Autopsy findings of the 1957-1958 influenza pandemic revealed that in 6 cases with influenza pneumonia, without secondary bacterial infection, there was is a fulminating diffuse haemorrhagic pneumonia which led to death in five patients. The trachea and bronchi contained bloody fluid and mucosa were hyperemic. Tracheitis, bronchitis and bronchiolitis with loss of normal ciliated epithelial cells were prominent and were frequently associated with evidence of epithelial regeneration. Submucosal hyperemia, focal haemorrhage, haemorrhage, oedema and a slight cellular infiltrate were present in these areas. The alveolar spaces contained varying numbers of neutrophils and mononuclear cells mixed with fibrin and oedema fluid. The alveolar capillaries were markedly hyperemic and intra-alveolar haemorrhage was common. Focal necrosis of the alveolar septa was found in only one case. In general, all changes described were most marked in the lower lobes. In each of the cases, striking acellular hyaline membranes lined many of the alveolar ducts and alveoli. Such hyaline membranes were frequently prominent in areas that showed minimal cellular exudation [24].
Limited autopsy findings of fatalities of the H5N1 Avian influenza viral epidemic of 1997 have been reported [25]. Although there were differences, compared to pathology reports referring to other strains of influenza, findings in the respiratory system in two cases revealed extensive haemorrhage, organizing DAD with interstitial fibrosis and cystic and dilated air spaces. Both cases showed a reactive hemophagocytic syndrome in hematopoietic organs, which the authors postulated may have been triggered by elevated reactive cytokines, i.e. the ‘cytokine storm’. H5N1 viral replication was not confined to the respiratory tract but also occurred in the gastrointestinal tract. However, together with alveolar macrophages, the type II pneumocytes were the major site of H5N1 viral attachment and replication in humans. The type II alveolar cells are important for surfactant production, fluid transport out of the alveolar lumen and re-epithelialization after damage, while alveolar macrophages are important for phagocytosis of pathogens and regulation of the inflammatory response in the alveoli [26].
Recently emphasis was placed on atypical presentations of COVID-19, including the ‘new’ finding of lung microthromboses and / or presence of pulmonary embolism [27]. However, the latter is not novel when one considers pathology articles published 100 years ago. Vascular injury in relation to viral (influenza) pneumonia was already described in 1919. LeCount reported hemorrhages and oedema in the lung, disseminated necrosis of the interalveolar capillaries and "button-like" firm peripherally located regions of consolidation ascribed to haemorrhagic infarcts, due to possible embolism [19].
Pathology findings in cases of the H1N1 influenza pandemic of 2009 predominantly showed DAD, accompanied by haemorrhage and necrotizing bronchiolitis [26]. The previously described three distinct patterns of pulmonary pathological changes were again found. These included classic exudative DAD, with alveolar and interstitial oedema, alveolar fibrinous exudate with hyaline membranes. Additional findings included microthrombi or thrombi in large arteries. Those cases dying within the first week after diagnosis presented with signs of exudative ARDS, whereas those dying after the first week presented with signs of proliferative ARDS. The case with the longest ICU stay showed fibrotic changes [28]. A detailed description of the histopathological changes and ultrastructural findings of a two fatal cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection were available for review [29,30]. In both cases the predominant pulmonary histologic pattern was exudative phase diffuse alveolar damage with denuding of bronchiolar epithelium, prominent hyaline membranes, alveolar fibrin deposits, type 2 pneumocyte hyperplasia, multinucleated syncytial cells, and alveolar septa showing oedema and lymphocytes with fewer plasma cells, neutrophils, and macrophages. Dispersed foci of necrotic debris were seen both sub pleural lung and within alveoli. In the one case the alveolar spaces were disrupted, dilated and contained a large amount of blood and fibrin, mixed inflammatory cell infiltrate and cellular debris [29]. Pneumocyte hyperplasia and reactive changes, denudation and sloughing of alveolar cells, rare multinucleated syncytial cells, congestion of the alveolar walls and hyaline membrane formation were evident. Lung vascular thrombi were not described.
SARS CoV-2 or COVID-19 histological examination of one case showed bilateral, diffuse alveolar damage with cellular fibro-myxoid exudates [31]. The right lung showed desquamation of pneumocytes and hyaline membrane formation. The left lung tissue displayed pulmonary oedema with hyaline membrane formation. Interstitial mononuclear inflammatory infiltrates, dominated by lymphocytes, were seen in both lungs. Multi-nucleated syncytial cells with atypical enlarged pneumocytes, characterised by large nuclei, amphophilic granular cytoplasm and prominent nucleoli were identified in the intra-alveolar spaces. The authors concluded that pathological features of COVID-19 greatly resemble those seen in SARS and Middle Eastern respiratory syndrome (MERS) coronavirus infection.
Pathologic findings from two patients included oedema and prominent proteinaceous exudates, vascular congestion and inflammatory clusters with fibrinoid material and multinucleated giant cells [32]. Reactive alveolar epithelial hyperplasia was seen in the one case and fibroblastic proliferation (fibroblast plugs) in the other,- in keeping with early organization. No prominent neutrophil infiltration was seen. Microvascular thrombosis was not reported.
Menter et al. studied autopsy findings of 21 COVID-19 patients in who the primary cause of death was respiratory failure. He demonstrated diffuse exudative alveolar damage with prominent capillary congestion, frequently accompanied by microthrombi despite anticoagulation [33]. The most prominent histologic finding was severe capillary congestion (capillarostasis) and the presence of microthrombi in the lungs and kidneys (despite anticoagulation), hyaline membranes, reactive pneumocyte changes and syncytial cells corresponding to exudative DAD. Ten cases showed superimposed bronchopneumonia. Further findings included pulmonary embolism (n=4), alveolar haemorrhage (n=3) and vasculitis (n=1). Pathology in other organ systems were predominantly attributable to shock and three patients showed signs of generalised thrombotic microangiopathy. A third of patients presented with severe mucous tracheitis/tracheobronchitis. Gross findings of the lungs were heterogeneous, ranging from patchy to diffuse areas of consolidation to severe and extensive suppurative broncho-pneumonic infiltrates. In all cases, the lung parenchyma was heavy and firm, unevenly blueish-red in colour with signs of severe congestion. Eight cases presented with proliferative DAD. Some cases had oedema and alveolar haemorrhage in conjunction with pulmonary embolism. In five of eleven cases where immunohistochemistry for fibrin were performed, microthrombi were detected in alveolar capillaries. Four cases presented with peripheral and prominent central pulmonary embolism. Direct correlation with radiology findings was not possible as most cases were only subjected to CT imaging at the time of hospital admission and not during the further course of their hospital stay. Ground glass infiltrates on chest X-ray were recorded in 57% of those subjected to X-ray examination.
The authors found that hypertensive, elderly, obese, male individuals with severe cardiovascular comorbidities, as well as those with blood group A, may have a lower threshold of tolerance for COVID-19. Evidence suggests that blood group A may be associated with the failure of pulmonary microcirculation and coagulopathies in COVID-19 and previous evidence investigating SARS-CoV suggests a direct interaction between blood group antigen A and the viral S protein, thus facilitating viral entry via ACE2. COVID-19 again emphasised the importance of virus-induced vascular dysfunction in disease progression. Notably, suppressing a COVID-19 associated “cytokine storm” by anti-interleukin 6 (IL-6) therapy now is a proposed strategy pursued in COVID-19 treatment [33].
Zhang et al. [34] described similar histopathological findings in cases of severe acute respiratory syndrome when compared to pathology findings in coronavirus disease 2019 (COVID-19).Diffuse alveolar damage (DAD) was a characteristic finding in non-survivors with both SARS and COVID-19. Patients who died less than 10~14 days of disease duration, demonstrated acute-phase DAD, while cases beyond 10~14 days of disease duration exhibited organizing-phase DAD in SARS. In addition, organization and fibrosis were usually accompanied by exudation. Coronavirus was mostly detected in pneumocytes and were not as prominent in macrophages and bronchiolar epithelial cells. Thrombosis was commonly observed in small vessels and capillaries in lungs in which the DAD pattern was noted. Microthrombosis was also found in extrapulmonary organs in COVID-19, but less than reported in SARS.
Haemorrhagic necrosis and lymphocytes depletion were found in lymph nodes and spleen in both SARS and COVID-19, suggesting pathological basis for the observed lymphocytopenia in some of the COVID 19 patients.
In an autopsy study of 38 COVID-19 related deaths from Northern Italy, the predominant findings were exudative and proliferative phases of DAD: capillary congestion, necrosis of pneumocytes, hyaline membranes, interstitial oedema, pneumocyte hyperplasia and platelet-fibrin thrombi in small arterial vessels were demonstrated. The inflammatory infiltrate included macrophages in the alveolar lumens and lymphocytes in the interstitium [35].
Ackermann et al. [36] compared the morphologic and molecular features of lungs obtained from autopsy of 5 unventilated and 2 ventilated patients who died from COVID-19, with lungs from patients who died from influenza A-related virus subtype H1N1 pneumonia (all ventilated) and age-matched, uninfected control lungs. The COVID-19 cases all died within 10 days of disease onset. A novel finding from the lungs of the patients with Covid-19 was that of vascular endothelialitis, thrombosis and angiogenesis, compared to the influenza and control cases. The lungs from the patients with COVID-19 and those with influenza shared a common morphologic pattern of DAD and infiltrating perivascular lymphocytes. All lung specimens from the COVID-19 group had DAD with necrosis of alveolar lining cells, type-2 pneumocyte hyperplasia and intra-alveolar fibrin deposition. Four of the 7 cases had focal changes and mild interstitial oedema. In three there were homogeneous fibrin deposits and marked interstitial oedema with early intra-alveolar organization. The severe changes to the endothelial cells in patients with COVID-19, suggested that the finding of SARS-CoV-2 virus within the endothelial cells, together with perivascular inflammation, contributed to the endothelial injury. Lungs of COVID-19 patients had widespread vascular thrombosis with microangiopathy and occlusion of alveolar capillaries. An unexpected finding was that of significant vessel growth (angiogenesis) in the lungs from patients with COVID-19 as compared with the lungs from patients with influenza. The specimens in the influenza group had florid DAD with significant interstitial oedema and extensive fibrin deposition in all the cases. The weight of the lungs of the influenza patients was significantly higher than that of the COVID-19 and control cases. There were no significant differences in the relative counts of ACE2-positive cells when comparing alveolar epithelial cells and endothelial cells of COVID-19 and Influenza cases. Analysis of precapillary vessels showed the presence of thrombi to a similar extent in lungs from patients with COVID-19 and lungs from the patients with influenza. Thrombi, without complete luminal obstruction, were consistently present in pulmonary arteries with a diameter of 1 mm to 2 mm. Fibrin thrombi of the alveolar capillaries could be seen in all the lungs from both groups of patients. Alveolar capillary microthrombi were 9 times as prevalent in patients with COVID-19 compared to patients with influenza when considering the mean number of thrombi per square centimeter of vascular luminal area. Intravascular thrombi in postcapillary venules of less than 1 mm diameter were seen in lower numbers in the lungs from patients with COVID-19 than in those from patients with influenza. Two lungs in the COVID-19 group had involvement of all segments of the vasculature when compared with four of the lungs in the influenza group. In three of the lungs in the COVID-19 group and three of the lungs in the influenza group, combined capillary and venous thrombi were found without arterial thrombi. The histologic findings of the lungs were confirmed by micro-computed tomography (CT). Lungs from patients with COVID-19 and from patients with influenza showed nearly total occlusions of precapillary and postcapillary vessels, lending support to a conclusion that microvascular vessel occlusion is not peculiar to COVID-19 as it already was demonstrated in in SARS CoV and post mortem studies from the previous century. The lungs from patients with COVID-19 however, had significant new vessel growth through a mechanism of intussusceptive angiogenesis. It is speculated that this is be related to a greater degree of endothelialitis and thrombosis.
In summary, whatever the origin of ALI or ARDS, it appears that histological studies demonstrated the presence of diffuse alveolar damage with hyaline membranes, interstitial oedema, cell necrosis and proliferation or fibrosis [21,37]. Lung histopathology after viral-induced ARDS, including coronavirus and COVID-19, are typically described as passing through three overlapping phases an inflammatory or exudative, a proliferative and finally a fibrotic phase (Figure 1). These phases may be complicated by episodes of nosocomial pneumonia and / or exacerbated by inappropriate mechanical ventilator strategies.
DAD is considered the pathological correlate of the clinical diagnosis of ALI/ARDS. Many authors suggest that the key finding that defines DAD is the presence of hyaline membranes. Hyaline membranes are characterized by the presence of dense eosinophilic material composed of cellular debris, plasma proteins and surfactant and are found along the alveolar septa. Fibrinous thrombi may be present in the capillaries of alveolar septa and alveolar ducts, as well as in small pulmonary blood vessels [37]. Possibly as a result of these thrombi, alveolar septa become necrotic. The late or chronic stage of influenza / viral ALI/ARDS is characterized by re-epithelization of the alveoli by type II pneumocytes (type II pneumocyte hyperplasia), squamous metaplasia and interstitial fibrosis of alveolar septa and microcystic honeycombing, a chronic condition analogous to ‘bronchopulmonary dysplasia’ described in ventilated, surfactant-deficient premature babies [21,29,33,37].
Although DAD is considered the pathological correlate of the clinical syndrome ALI/ARDS, clinicians have recently proposed that COVID-19 cases often presents with as an ‘atypical’ format, i.e. dissociation between degree of hypoxaemia, respiratory rate (as indicative of respiratory distress) and lung compliance [16-18,20,37,38]. This observation begs the question whether this is a novel manner in which COVID-19 presents? [4]. In 2017 it was already reported that clinical and autopsy studies suggest that only one-half of patients who meet the clinical definition of ARDS have DAD [37]. The authors then questioned whether patients with ARDS who have DAD, experience different outcomes than patients with ARDS but without DAD? [37]. It appears therefore that ‘atypical’ ALI/ARDS is not uncommon, neither unique to COVID-19. Alternatively, this dissociation between the hypoxemia and respiratory distress may be an earlier expression and focal lung injury compared to the later general inflammatory process driven ALI/ARDS involving the whole lung.
ALI/ARDS is caused by conditions which either directly affect the lung (pneumonia, aspiration of gastric contents, blunt chest trauma) and non-pulmonary causes such as trauma, sepsis, acidosis, prolonged haemorrhagic shock and many other conditions [39-42]. The final common pathway by which these diverse aetiologies injure the lung is via activated neutrophils which accumulate in the lung vasculature and move into the lung tissue per se [41,43,44]. In this regard the lung is only but one of the organs involved in what is known as multi-organ dysfunction and failure (MOD/F) [45]. ARDS develops rapidly, in most patients within 12 to 48 hours of exposure to infectious or non-infectious insults [44]. Every component of the lung (epithelium, endothelium of the vasculature, airspaces and interstitium) are involved and this process [45].
Characteristically, there is a latent period between the insult and the development of the full-blown clinical syndrome ALI/ARDS. After this interval; tachypnea, laboured breathing and cyanosis (hypoxemia) are observed. The major insult is to the alveolar-capillary membrane that initially results in increased permeability and subsequent interstitial and alveolar haemorrhagic pulmonary oedema. This early stage is followed by an inflammatory process and the formation of hyaline membranes to be followed by fibrosis and perhaps recovery of the lung architecture [46]. This course of events is also described as the diffuse alveolar damage (DAD) syndrome [21,37,38,47]. The injury of the alveolar-capillary membrane eventually causes protein-rich neutrophilic exudate in the alveoli (although alveoli are involved in a seemingly random manner as there is variation in the alveolar injury ranging from limited injury to severely injured alveoli (Figure 2).
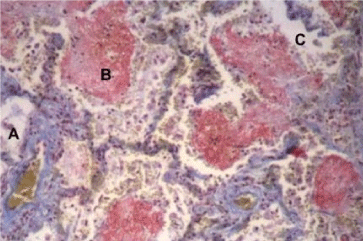
Figure 2. Histology of lung of a patient who died with ARDS. Note the differential distribution of the alveolar abnormalities (compare A, B and C). (A Coetzee, N. Rossouw, unpublished result).
This process, combined with a defective hypoxic pulmonary vasoconstrictive response, results in low ventilation/perfusion units (Qva and Qs) and arterial hypoxia [48]. It has been shown that the dead space (VD/VT) is also increased [49] and the likely explanation is the obstruction of the pulmonary artery by clots and obstruction of the arterioles. Hence, the probable cause for the dead space is more likely the vascular pathology rather than lung parenchymal pathology (which explains the low ventilation to perfusion pathology) [50]. It needs to be emphasised that ALI/ARDS is not only a lung parenchymal disease, the pulmonary vascular bed are involved and increased clot formation is very much part of the process and at least partially explains the acute pulmonary artery hypertension which may fail the right ventricle and result in an incompetent cardiac output [45,51]. The reduction in cardiac output occurs when the thin walled right ventricle cannot cope with the acutely elevated pulmonary artery pressure and this interaction results in a mismatch between the oxygen delivery and consumption because of the resultant insufficient cardiac output. The latter results in mixed venous blood desaturation and, in the presence of Qs, the mixed venous desaturation is a significant contributor to arterial hypoxia (Figure 3).
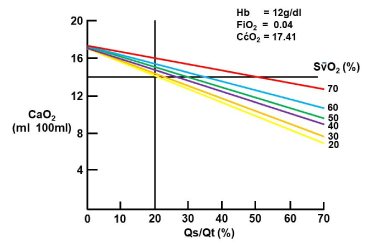
Figure 3. The important role of mixed venous saturation on arterial oxygenation in the presence of pulmonary shunt. The mixed venous are shown on the right. (Computed from 46 patients with ARDS and using the average values as indicated on the figure (A Coetzee, Unpublished result, 1988).
The hypoxia associated with the latter process cannot be overcome with ventilator strategies; it requires inotropic support of the right ventricle and an increase in the arterial oxygen content for instance by ensuring an acceptable haemoglobin value [51]. This well described vascular pathology, and its effects on the central circulation and arterial oxygenation, is the basis for the concept of nebulized heparin and thrombolysis [52]. The pathophysiology of the ALI/ARDS characteristically results in a reduction in lung compliance by virtue of the reduction in the surfactant and function residual volume (FRC), irrespective if one supports the contention of whole or regional (or both) lung pathology [42,53,54]. The regional model is referred to as the “baby lung” concept associated with ALI/ARDS and was the major driver for the protective lung ventilation strategy [54,55].
Among infectious organisms, viruses can produce the characteristic acute phase pathophysiologic hallmark of ARDS, i.e., DAD [37,56,57]. Failure to rapidly overcome or repair the tissue damage results in a negative spiral of self-perpetuating inflammation with subsequent progressive loss of lung parenchymal function, intravascular clotting and right heart failure [38,45,47,51,55,57-59]. Since ARDS was first named and defined in 1967 (it probably was already described in 1945), the definition has been redefined several times [38,58]. However, the described pathology remained unchanged [59,60]. Ashbaugh et al. described acute respiratory distress (ARDS) in 12 patients with acute onset of tachypnoea, hypoxaemia, and loss of compliance after a variety of insults. In 4 of the cases, viral pneumonia was considered as the underlying cause [60]. The clinical and pathological and X-ray features closely resembled those seen in infants with respiratory distress, congestive atelectasis and post-perfusion lung [61,62]. Positive end-expiratory pressure (PEEP) improved the atelectasis and relieved the hypoxaemia [63]. Autopsy in 7 patients showed heavy and deep reddish-purple lungs, resembling liver tissue. In those who died early in the course of the illness, predominant features were hyperemia, dilated engorged pulmonary capillaries, and areas of alveolar atelectasis, interstitial and intra-alveolar haemorrhage, pulmonary oedema and hyaline membranes. Alveolar macrophages were numerous. No vascular thromboses were however reported. Lung surface-tension was found to be high in two cases. Diffuse interstitial inflammation and fibrosis, without notable hyperaemia, were present in two patients who died after a protracted course. Both patients also had hyaline membranes.
Classically, ARDS is recognized to be a neutrophil-driven disease but experimental data have shown that even neutropenic patients are susceptible to the development of ARDS [38,41]. In addition, the involvement of cells from the innate (including macrophages and platelets) and adaptive immune systems in the pathogenesis of acute respiratory distress syndrome are increasingly recognized. Neutrophils and macrophages are recruited to the inflammatory focus on the lung, thereby propagating the initial insult. The inflammatory exudate interacts and decreases the function of surfactant, causing alveolar instability and as the epithelial Type 2 alveolar cell injury progresses. The presence of hyaline membranes and fibrin clots within the distal- and alveolar spaces, alter the lung viscoelastic properties leads to decreased pulmonary compliance and abnormal gas exchange via ventilation to perfusion mismatching and ineffective hypoxic pulmonary artery vasoconstriction response [38,48]. Pulmonary macro and microvascular thrombi occur and probably explain to a large extent the acute pulmonary artery hypertension which increases right ventricular afterload [45]. The right ventricular dysfunction and ventricle-afterload mismatch can be further exacerbated by incorrect mechanical ventilation strategies and excessive fluid administration [61].
Systemic elevation of cytokines is involved in the pathogenesis of ARDS-related morbidity [62]. Persistent production of inflammatory mediators in the lung sustains inflammation with resulting tissue injury, intravascular and extravascular coagulation and fibroproliferation. These result in maladaptive lung repair ultimately evolving into fibrosis. In COVID-19, the pattern of immune dysregulation is characterized by IL-6-mediated human leukocyte antigen D related (HLA-DR) expression and lymphopenia. This causes with a sustained cytokine production and hyper-inflammation [63].
At a clinical level, ARDS is defined as the presence within 1 week of a known clinical insult, acute arterial hypoxemia (PaO2/FiO2 ≤ 300 mmHg) with a minimum requirement of 5 cm H2O positive end-expiratory pressure (PEEP) and the presence of bilateral radiographic opacities not explained by an elevated left atrial pressure [58].
Clinical experience is in keeping with the well described pathology. Initially the patients have signs and symptoms which can be ascribed to lung oedema i.e., degrees of hypoxemia and tachypnea, mainly driven by the hypoxia and the loss of compliance associated with a reduction in functional residual capacity and the associated stimuli originating from the lung volume receptors. As the disease progresses, the hypoxia deepens, and the pathophysiology of the hypoxia is the low ventilation -perfusion (V/Q) units varying from venous admixture (Qva) to complete pulmonary shunt (Qs). The mixed venous blood oxygenation becomes more important in maintaining arterial oxygenation as the Qs/Qt increases. With regards to the latter, the clots demonstrated in the pulmonary artery and perhaps some vasospasm elevates the pulmonary artery pressure, and this acutely loads the thin-walled right ventricle [51]. This mismatch in right ventricular and pulmonary artery elastance reduces the mixed venous saturation and contributes significantly to the arterial hypoxia. The low pulmonary compliance is reflected in the accompanying need for increased inflation pressure required to ventilate the patients. However, the importance of containing the inflation pressure and driving pressure, in order to protect the lung, has recently been repeatedly emphasized [64-66]. In order to maintain the plateau inflation pressure below 30 cm H2O, rather than risking volume trauma of the lung, the concept of permissive hypercarbia was promoted and has been shown to be safe [67].
Recently it was reported that the pulmonary pathology associated with COVID-19 cases with respiratory involvement, vary from minimal (acute lung injury) to severe lung involvement (ARDS) [4]. During the early phase of ALI, patients developed severe hypoxemia often associated with near normal respiratory system compliance [68]. The early CT scans showed patchy infiltrates and apparently normal lung volumes [27]. The proposed mechanism for the hypoxia is via the inhibition of the hypoxic pulmonary arterial vasoconstrictive response (related to inflammation and associated tissue injury) which results in these areas acting as low V/Q lesions. We speculate when we propose that the absence for overt tachypnea, despite severe hypoxia, is at least partially the result of the well-maintained lung volume and hence the absence of a volume receptor driven fast respiratory rate [68]. This early presentation is different from the more usual presentation of ALI /ARDS where the patients have a significant tachypnea to the point of mechanical respiratory failure [69]. Already in 1971, Petty and Ashbaugh referred to the hypoxia and mechanical ventilatory stress when they reported that ‘Patients with this syndrome suddenly develop marked tachypnea, dyspnea and cyanosis which is refractory not only to nasal oxygen but also to intermittent positive pressure breathing’ [59]. This atypical pattern of COVID-19 presentation could still be classified under ALI given that the lung involvement may only be present in quadrants and not meet the bilateral involvement required to meet the definition of ARDS [8,58].
COVID-19 is associated with an accelerated inflammatory course (hyperactivation of monocyte-derived macrophages) that is associated with a coagulopathy [70]. It was suggested that the dissociation between oxygenation status and lung mechanics in patients with COVID-19 pneumonia, is an atypical form of the condition and that micro- and macro-thromboses in the lung contributed to the ventilation-perfusion mismatch [4,5]. However, on a physiological basis, the vascular occlusion results in pulmonary dead space (high ventilation to perfusion ratios) [50]. The latter, in principle, cannot explain hypoxia save for a (initially) elevated CO2 which, according to the alveolar gas equation, can depress the alveolar oxygenation in a 1.2 ratio (accepting the respiratory quotient being 0.8). This (initial) increase in PaCO2 associated with a dead space lesion, will drive the respiratory centre and, as VA = VE – (1-VD//VT), the increase in minute ventilation can usually overcome the hypercarbia (within limits). The hypoxia must have another origin (i.e., cannot physiologically be explained by the micro clots in the lung vasculature). From a pathophysiological perspective the combination of low ventilation perfusion alveolar units and loss of the hypoxic pulmonary vasoconstriction explains the hypoxia seen in COVID-19. The loss of the pulmonary hypoxic vasoconstriction mechanism, which usually serves to optimize the ventilation to perfusion in the lung, is inhibited by active infective processes and injury and there is no reason to suspect that it would be different in viral infection(s) [71-74].
The above raises the question whether vascular thrombosis is a new and or a unique finding specific to COVID-19 infection? Although alveolar capillary microthrombi appears to be 9 times as prevalent in patients with COVID-19 compared to influenza A cases [36], review of pathology findings suggests that lung vascular thrombosis is not a new phenomenon and that micro and macro pulmonary vascular thrombosis / thromboembolism is an integral, but perhaps previously underestimated and unrecognized, aspect of the pathophysiology of most viral-induced ALI/ARDS as well as in ARDS from other causes [45]. Before COVID-19, Thille et al. found that 24% of the 159 overall populations with direct or indirect ARDS had thrombosis of small pulmonary vessels at autopsy examination [20]. In our experience with ARDS, clotting appears to be an early event in the disease process [74]. We therefore suggest that there is no need for a name change of COVID-19 to MicroCLOTS (microvascular COVID-19 lung vessels obstructive thrombo-inflammatory syndrome), - a proposed new name. The pulmonary vascular lesions noted in COVID-19 are, as a fact, not a new phenomenon [6].
On the basis of their interpretation of COVID-19 related hypoxemic respiratory decompensation, Gattinoni et al. then proceeded to differentiate between ‘ARDS’ phenotypes [4,5]. They suggested that an initial (early) presentation could be described as Type L (or ARDS 1) because it is characterized by low elastance (i.e., high compliance), low ventilation to perfusion ratios, low lung weight and low recruitability. Type H (later) or ARDS 2 has high elastance, high right-to-left pulmonary shunt, high lung weight and high recruitability. ARDS 1 / Type L may progress to ARDS 2 / Type H. It is therefore recommended that respiratory supportive management should also be adapted according to the phenotypic type.
Individuals afflicted with ARDS associated with COVID-19 often suffer from a range of other underlying disease subtypes (phenotypic heterogeneity), which increases morbidity and mortality. Individuals may also present at different stages of a dynamic disease process (temporal heterogeneity). It is therefore understandable that no single approach predicts or illuminates the possible clinical course which will be followed. During the initial Type 1 / ARDS 1 phase of COVD-19 infection, it is suggested that CPAP, heated, humidified, high flow oxygen (HHHFO2) or other forms of non-invasive ventilator (NIV) support could be utilized since the pathophysiology suggests a better preserved functional residual capacity (see CT scans reference [27]). We therefore speculate that CPAP may not be as effective as one expects as the areas of the lung, which is unaffected by the focal areas of pathology, probably has better compliance than the diseased areas. Hence the PEEP will preferentially benefit the more “normal” lung and not specifically the diseased areas. However, early recognition of the change from Type 1 to Type 2 is necessary as Type H / ARDS 2 require timely intubation and application of formal mechanical assistance and titrated PEEP since these cases, in addition to severe hypoxaemia, have low lung compliance.
In early ALI associated with COVID-19, the management of the hypoxic patient is recognizing progression of Type1 to type 2 ARDS requires assessment of the respiratory mechanics (as expressed by a reduction in lung compliance and associated increased respiratory rate) and pulmonary parenchymal dysfunction and failure (type I respiratory failure). The latter is usually gauged from the arterial oxygenation and or the calculation of pulmonary shunt or the derived indirect indices of shunt (PaO2/FiO2, Alveolar-arterial oxygen gradient [AaDO2]). However, although the indirect indices of shunt are commonly used, the latter does not reliably represent pulmonary shunt [61]. Probably a more accurate indirect estimation of pulmonary shunt can be obtained from the (linear) equation Qs/Qt (%) = 88.77- 48.96 (logPaO2/FiO2) (PaO2 in Kpa) (Coetzee A, unpublished data).
A factor which perhaps complicates the clinical course of viral-induced ARDS is related to patient’s breathing efforts [15]. Gattinoni et al. postulated that progress from high to low compliance lungs are due to the natural course of COVID-19. However, they could not exclude the possibility that lung oedema was in part due to the initial respiratory management of patients who presented with high respiratory drives and vigorous inspiratory efforts. The latter resulted in high negative intrathoracic pressures and the authors suggested that, in addition to viral pneumonia, these patients are likely to follow a more severe course due to, inter alia, a self-inflicted (S) (patient) lung injury (P-SILI). This category should best respond to the timely application of invasive mechanical ventilatory support in order to limit or prevent the transition from Type L to H [5].
P-SILI has been demonstrated in an animal model of stimulated hyperventilation and is now recognized as a contributory factor to the spectrum of acute lung parenchymal injury [75]. In this regard, Wolbach reported interesting autopsy findings in cases of the 1918 influenza pandemic. The author described the presence of air in the subcutaneous area in the neck tissues and, in some cases, over the entire trunk, head and extremities. He speculated at the time that the acute interstitial emphysema resulted from the mechanical rupture of some of the pulmonary alveoli which have become distended by acute alveolar emphysema (a negative pressure volotrauma) [15]. Although not recognized as P-SILI at the time, it is perhaps appropriate to speculate that this was a likely contributory factor, since no patient was ventilated. Underlying lung pathology in those cases revealed alveolar emphysema with accompanying haemorrhagic exudate, in some instance’s interstitial emphysema, and the formation of hyaline membranes in the alveoli and the alveolar ducts. Early ‘virus’ associated lung lesions consisted of injury (desquamation) to the epithelial cells of the airways, alveolar ducts and the lining of the alveoli. At the same time there was congestion of the blood vessels in these areas and presence of red blood cells, fibrin and hyaline-like membranes in the alveolar spaces were noted. In places, the alveolar spaces were markedly distended with air, so that adjacent lung tissue was compressed. It was assumed that the haemorrhage and hyaline material in some way acted as ball-valve effect causing air trapping, alveolar over distention and air leaks. It is speculated in some cases lung injury was caused by increased tidal volumes as may have occurred during forceful spontaneous breathing initiated by a high respiratory drive. This in turn, lead to lesions that appear similar to the ventilator induced lung injury (VILI) later observed in mechanically ventilated subjects [75]. This injury would be similar to volutrauma, albeit the alveolar distention was caused by negative pressure rather than positive pressure insufflation [76].
Although there are more sophisticated surrogate markers of worsening ALI/ARDS in the clinical setting, excessive inspiratory effort (reflecting the increased work of breathing due to the stiff lung), PaO2/FiO2 (measured within the first 6 hours after hospital admission), dyspnea or response to non-invasive breathing support (CPAP, heated humidified high flow oxygen [HHHFO2]) or non-invasive ventilation (NIV), could be considered as markers of worsening respiratory distress or increased lung stiffness and the developing of ARDS 2 / Type H [4,38- 40,77].
It appears that there may be windows of opportunity for selective therapeutic and supportive interventions during disease progression. Benefit may also depend on the severity and nature of the underlying cause of respiratory dysfunction or failure.
In the mechanically ventilated adult with ALI/ARDS, there are now considerable experimental and clinical evidence showing that the application of high levels of PEEP in the initial phases protects against alveolar stress and support gas exchange by maintaining collapsed alveoli open hence improving functional residual capacity and pulmonary compliance [78]. However, in the presence of surfactant insufficiency, the overall effects of PEEP on ability to recruit and maintain patency of alveoli in ALI/ARDS patients are complex. The percentage of lung which can be recruited, analysed with CT scans performed up to 45 cm H2O applied airway pressure, varied between patients [79]. In adult patients with acute respiratory failure, non-invasive ventilation (NIV) with positive-pressure delivered through a face mask, was as effective as conventional ventilation in improving gas exchange and was associated with fewer serious complications, shorter intensive care unit stay and survival [80]. However, when considering the results, one need to take cognisance of the fact that 10 of the initially non-invasive patients had to be intubated and the shorter ventilation time and improved survival could also indicate that the patients subjected to NIV was perhaps less ill. Given the random allocation to both NIV and conventional ventilation, one has to query the power of the results. The more sick patients were moved from NIV to conventional ventilation i.e. already an indication of bias.
Several studies reviewed or evaluated a role for NIV in patients with hypoxemic respiratory failure [81-84]. Although one review reported that benefit may only be possible in a few selected patients [82], the evidence is accumulating to show that among patients with hypoxemic respiratory failure or ARDS, use of NIV with various interfaces reduce the risk of tracheal intubation and / or impacted on survival [81,83,84]. This suggests that the loss of alveolar stability is an important factor in the progress of the pathophysiology inasmuch as NIV, and even high-flow nasal oxygen (HFNO), allows the ability to supply PEEP (directly and indirectly) and hence affect alveolar stability. This raises the question whether NIV and HHFNO with additional surfactant treatment should not be considered to stave of invasive ventilation and its associated risks?
In this regard, is surfactant a potential adjunctive treatment that is missing from the equation when NIV or CPAP are applied in patients with ALI/ARDS? New techniques of surfactant administration in combination with CPAP in adults may offer a manner to avoid invasive mechanical ventilation. The combination of non-invasive breathing support (CPAP) or humidified heated high-flow nasal canula oxygen (HHHFNCO2) and early surfactant treatment has been shown to be effective in recruiting lung volume in surfactant deficient premature newborn infants and in ventilated patients with ALI/ARDS, whereas recruitment manoeuvres without surfactant resulted I poorer outcomes [85]. A probable explanation is that, in the absence of PEEP/CPAP, there is selective distribution of ventilation to alveoli already ventilated and the collapsed alveoli not having the benefit of surface tension stabilization associated with instilled surfactant [86]. The proposed advantage is that either CPAP or PEEP recruits alveoli and, if surfactant is then instilled, it will assist in maintaining the alveolar volume (in conjunction with the mechanical support of CPAP or PEEP). Similarly to the non-invasive strategies followed in nRDS, adult ALI/ARDS could perhaps be treated with NIV/CPAP in combination with early rescue exogenous surfactant during the early phase of the disease. A recent trial of exogenous surfactant treatment in a group of adult patients diagnosed with direct or indirect ARDS, revealed improvement in the oxygenation status and duration of ventilation in the group treated with porcine derived surfactant when compared to controls [87]. Although the quality of the study is uncertain and the trial methodology and randomization difficult to extract from the publication, it appears that the authors instilled surfactant according to the less invasive management strategy already adopted by neonatologists (‘Intubation-Surfactant-Extubation’ (InSurE) approach) [88]. Brief tracheal intubation, followed by surfactant administration and then extubation of preterm neonates with RDS, who are then stabilized on CPAP, has revolutionized management and decreased mortality without resulting in deterioration of other outcomes [89]. It may be worthwhile considering following in the example of the neonatology practice in adult patient with early ALI/ARDS.
Acute lung injury / ARDS is a complex clinical condition and, although on a pathophysiological basis there initially was an expectation that exogenous surfactant may assist in the management of the ALI/ARDS patient, it was always somewhat optimistic to expect that correcting one part of the pathophysiology, would necessarily translate to improved survival. However, in terms of management, it would be more appropriate to critically evaluate oxygenation and lung compliance as focussed outcomes for a proposed strategy of using exogenous surfactant in ALI/ARDS.
Despite the discouraging results reported in the use of surfactant in ALI/ARDS, we agree with other researchers that the ‘surfactant in ARDS’ concept should not yet be discountedsince other surfactant preparations and more efficient methods of delivery justifies the ongoing interest . Seeger et al. emphasized that pulmonary s
The major pathophysiology in nRDS is surfactant deficiency. If the experience in treating nRDS is taken as the standard, then early selective treatment in adults with exogenous surfactant, after stabilization on some form of continuous positive airway pressure (CPAP), should perhaps be reconsidered to improve short- and long term outcomes in adults [92,93].
In term infants and young children with severe direct acute lung injury, as a consequence of meconium aspiration (MA) or viral pneumonia, administration of exogenous mammalian-derived surfactant resulted in improved oxygenation and marginal clearing of opacities on the chest X-ray [94]. Several studies involving term infants with meconium aspiration have collectively shown that mammalian-derived surfactant treatment improved oxygenation and lung mechanics, reduced incidence of pneumothoraxes, decreased duration of mechanical ventilation and oxygen therapy, reduced time of hospitalization and reduced the need for ECMO [95-97].
Surfactant therapy also has clinical benefits in young infants and children one week to 2.5 years of age diagnosed with acute respiratory failure from respiratory syncytial virus (RSV) bronchiolitis infection. A recent meta-analysis of three studies, evaluating the use of surfactant therapy in the management of bronchiolitis in critically ill infants, concluded that surfactant had positive effects on the duration of mechanical ventilation, time spent in the intensive care unit, oxygenation and CO2 elimination [98,99].
A multicentre randomized trial of calfactant in infants, children and adolescents with ALI/ARDS (tracheal instillation of 2 doses of 80 mL/m2 calfactant; 35 mg/mL of phospholipid suspension in saline), reported improved oxygenation and a significantly reduction in mortality across the age groups [91]. However, there was no significant improvement in the course of respiratory failure measured by duration of ventilator therapy, intensive care unit or hospital stay. Twenty six percent of the enrolled patients were below 12 months of age. The trial reported minimal adverse events associated with surfactant treatment inter alia hypotension in 9% of the reported cases.
Although there are clear and important differences, the early course of ALI/ARDS does share some features with neonatal respiratory distress syndrome (nRDS) and neonatal ARDS (meconium aspiration). The similarities mainly refer to the pathophysiology, ventilation, diffusion, and perfusion and include surfactant insufficiency/deficiency, lung inflammation and alveolar epithelial injury. It also includes increased vascular permeability, heterogeneous alveolar atelectasis and consolidated lung regions resulting in low ventilation to perfusion lung units, lung oedema, and progressive deterioration in FRC. Finally, the neonates and adults share a reduction in lung compliance and pulmonary artery hypertension when they develop acute respiratory distress [85,100,101].
The early and late abnormalities of the alveolar surfactant system in the pathogenesis of ARDS in adults were reviewed by Seeger and co-workers almost 30 years ago [91]. The composition and total volume of lung surfactant in normal humans is constant between the ages of 13 months and 80 years. It changes in lung disease states [102]. Surfactant lipid is estimated to be at a concentration of 35-50 mg/ml. consisting of about 80% phosphatidylcholine. About 50% of the surfactant phosphatidylcholine by weight consists of the disaturated phospholipid dipalmitoylphosphatidylcholine (DPPC). Other PLs, such as phosphatidylglycerol (PG), and phosphatidylinositol (PI), as well as cholesterol (the major neutral lipid in surfactant), assists with adsorption, spreading, and fluidity of the surfactant film [103]. In addition, surfactant contains different apoproteins, neutral lipids, and carbohydrates. There is evidence that PG can suppress viral (RSV, Influenza A) infection and inflammatory responses in the lung [104-106]. PG and PI block recognition of activating ligands by the TLRs, either directly or via the TLR4 co-receptors CD14 and MD2 [106]. Whether a PG-containing surfactant will block CD14-mediated cellular activation in patients early in the development of ARDS in general and specifically due to SARS-CoV-2 infection, is unknown.
Rebello et al. reported that the adult human lung contains about 28 µ mol/kg body weight Sat-PC, and approximately 2 µmol/kg Sat-PC (7%) in the alveolar wash. The authors calculated that this pool size of 2 µmol Sat-PC/kg is equivalent to approximately 4 mg/kg surfactant [102]. Moreover, by using estimates of Brown et al of alveolar surface area at functional residual capacity (FRC), [107] and values for the molecular area of saturated-PC at low surface tensions in vitro (according to Watkins [108]), Pre and co-workers calculated a theoretical minimum amount of saturated-PC required to form a monomolecular film over the alveolar surface at FRC, is 2.97 mg/m2 [109]. With this insight, the alveolar pool size of surfactant in the adult human, based on five sub-segmental lavages of volunteers, was estimated as a mean of 1.4 mg Sat-PC/kg body weight, or about 3 mg surfactant/kg [109]. The suggested dose per kg should therefore theoretically correlate with the amount of surfactant required to cover the complete alveolar surface [109]. In view of the encouraging results from early clinical trials of exogenous surfactant therapy in acute lung injury [110], Rebello et al. concluded that when one considers the relatively small adult alveolar surfactant pool size (2 µmol Sat-PC/kg; 4 mg/kg body weight), that surfactant doses used in certain clinical trials, were excessive since it significantly exceeded the endogenous alveolar pool [102].
Since the initial report on ARDS by Ashbaugh et al. [60], biochemical dysfunction and abnormal biophysical behaviour of surfactant during the course of ARDS, have received continuous attention [110-115]. Surfactant abnormalities have been described in ARDS and changes in PL composition may last for weeks. Generally the recorded abnormalities are low PI, low PG, and low plasma myoinositol [111]. In ARDS, the main biochemical abnormalities of surfactant include an 80% decrease in the total phospholipid content, decline in the fractional content of DPPC and PG, large surfactant aggregates, and loss of apoproteins (90% of surfactant protein (SP-A and SP-B)) [103,116,117].
This loss of alveolar surfactant is the result of several factors including decreased release of surfactant by injured alveolar type 2 (AT2) cells and presence of alveolar plasma proteins secondary to influx of protein-rich alveolar oedema fluid associated with increased permeability of the alveolar-capillary membrane. Protein leakage into the alveolar space precedes surfactant abnormalities in patients during the course of post-traumatic ARDS [118]. Further loss of surfactant is due to cleavage of phospholipids by serum phospholipases, damage to surfactant compounds by inflammatory mediators (cytokines, chemokines, and secretion of proteases, as well as concomitant collagen synthesis), conversion to non-functional surfactant and incorporation of surfactant phospholipids and apoproteins into polymerizing fibrin when hyaline membranes are formed [91,119]. These fibrin-rich exudates in the alveoli are caused by the alveolar-capillary damage and are aggravated by inflammation related activation of blood coagulation and inhibition of fibrinolysis [91,120]. Incorporation of surfactant in fibrin-hyaline membranes results in intra-alveolar accumulation of clot material [91].
Surfactant dysfunction occurs early after the onset of direct ARDS. In one study, median time from diagnosis of ARDS to initial brocho-alveolar lavage (BAL) was 12.1 ± 1.3 hours and analysis of the BAL fluid revealed significant alterations of the surfactant kinetics [121]. There was a 10-fold reduction in phospholipid-to protein ratio (indicating leakage of proteins into alveolar spaces), a reduction in the amount of large surfactant aggregates (LA), decreased PC and PG, increase in the phosphatidylserine (PS), PI, phosphatidylethanolamine (PE), sphingomyelin (SPH) and a significant loss of all surfactant proteins (SPs). The hydrophobic surfactant proteins SP-B and SP-C, as well as SP-A, but not SP-D, were reduced. Within the PC fraction of the multi-lamellar aggregates (LA), a more than 50% reduction in DPPC was observed when compared to values obtained from controls. This was paralleled by a marked increase in unsaturated species of surfactant. Inactivation of intra-alveolar surfactant includes increased conversion of surface active large multi-lamellar aggregates (LA) to small aggregates (SA) with poor surface activity.
As a consequence of the above surfactant changes, the surface tension in the alveoli was increased. The abnormal lining of the alveoli increases retractile forces (increased surface tension) lead to fluid accumulation and protein leakage into alveoli and a decrease in compliance of the lung [111,114,116]. Early alterations in the composition of surfactant have also been described in patients at risk to develop ARDS [114]. In patients at risk of ARDS, BAL revealed a reduced surfactant pool because of decreased PLs. In patients at risk of ARDS (within 13±10 hours of ventilation) there was a 2-fold increased minimum surface tension (MST) and in patients with ARDS (within 87 ± 9 hours of ventilation), a 4-fold increase in MST, when compared to control patients. The “at risk” group was intubated, mechanically ventilated, and had at least one underlying predisposing risk factor for ARDS.
Interestingly, and perhaps in keeping with reports of a ‘new’ dissociative manifestation of certain COVID-19 ARDS cases [122], data has shown the total surfactant pool in patients demonstrated an increased level of lysophosphatidylcholine (LPC) in the at risk ARDS group [114]. Type-II secretory phospholipase A2 (sPLA2-II) plays a major role in the hydrolysis of surfactant phospholipids and its expression is inhibited by surfactant [123]. We speculate that this observation could be linked to the CT scan images and clinical presentation of the atypical COVID-19 patients inasmuch as they have relatively normal lung volumes and perhaps reasonably normal lung compliance. The associated, and somewhat out of context severe hypoxia which was noted, probably was the result of the defective hypoxic vasoconstrictive response (and in principle this does not primarily affects the lung volume and neither the lung compliance).
Niewoehner et al. [124] postulated that excess phospholipase A2 (PLA2) activity may be responsible for the formation lysophosphatidylcholine as a naturally occurring product of enzyme activity. Moreover, many snake venoms act through diverse enzymatic activities including phospholipase A2 activity, which catalyses the hydrolysis of the two acyl groups in sn-3 phosphoglycerides. This led Niewoehner and co-workers to investigate the direct effect of sPLA2 on the catabolism of pulmonary surfactant. In their experiments they incubated calf-lung surfactant with sPLA2 from the venom of the snake, Naja naja. This led to a marked decrease in the relative amounts of phosphatidylcholine and a parallel increase in the level of lyso-phoshatidylcholine. The authors then showed that intratracheal administration of PLA2 to the lungs of guinea pigs induced acute lung injury [124]. However, they concluded that extrapolation of their experimental results could not be made to mammalian sPLA2-II inasmuch as humans has less ability to hydrolyse the main surfactant phospholipid dipalmitoyl phosphatidylcholine (DPPC).
In experiments by Kakuta et al. it was demonstrated that artificial surfactant, consisting of DPPC, unsaturated phosphatidyl-glycerol and tripalmitin (65:25:10, w:w:w), enhanced ciliary beat frequency and accelerated recovery by reversing epithelial injury caused by hydrogen peroxide. In addition, the effect was DPPC dose dependent and ion transport augmented the effect. The authors concluded that a likely explanation for this phenomenon was that surfactant changes the rheological properties of the periciliary fluid [125]. This is perhaps another positive factor to consider when discussing surfactant therapy in lung disease.
Surfactant alterations occurring early during the clinical course of exudative DAD therefore ties in with the underlying pathophysiology (Type 1 and 2 epithelial- and vascular endothelial injury) and accompanying findings of increased interstitial, septal and intra-alveolar fluid accumulation, formation of distal airway and alveolar hyaline membranes, fibrin clots, atelectasis, and low ventilation to perfusion alveolar units. The latter results in a decrease in arterial oxygen tension. The loss in surfactant and functional residual capacity (FRC) result in a progressive decrease of lung compliance, increased work of breathing and ultimately, mechanical respiratory failure. The capillary and arteriolar occlusions cause pulmonary dead space lesions (being high ventilation to perfusion units) with an initial primary effect on carbon dioxide homeostasis and minute volume. The associated increase in dead space forces a further increase in minute ventilation which will further increase dead space given the interaction between the fixed anatomical dead space and decreased tidal volume associated with a tachypnea.
An interesting question is whether exogenous surfactant could be administered early during the course of developing ALI/ARDS in order to mitigate the risk of developing P-SILI in patients? As ALI/ARDS progresses, spontaneously breathing patients usually have a high respiratory drive (and increased negative intrathoracic pressure associated with the reduced compliance). This could perhaps lead to the development of P-SILI.
Despite still many unanswered questions and even reported drawbacks, some studies have shown functional benefits associated with the use of surfactant in adult patients with various degrees of ARDS [126].
Surfactant is integral to viscoelastic properties of the lung. It lowers surface tension and allows collapsed alveoli to open at lower inspiratory (inflation) pressure and maintain alveolar dimensions once inflated. This inter alia translates to the maintenance of an effective functional residual capacity (FRC) which is paramount in the maintenance of arterial oxygenation. Because of the surfactant deficiencies/insufficiencies and airway obstructions due to fibrin-rich clots, especially in dependent or lower regions of the lung, patients suffering from ALI /ARDS are not able to adequately maintain their FRC [127].
Surfactant replacement in adults with ARDS has resulted in a temporary improvement of gas exchange properties [117]. It is therefore reasoned that there could be a role for exogenous surfactant replacement treatment during the early phase (Type 1) of direct ALI/ARDS, inter alia caused by viral pneumonia. Administration of surfactant earlier in the course of the disease, when lung inflammation is present, but before severe lung dysfunction occurs, may perhaps be of value in the limiting the subsequent extent of the required management of the lung dysfunction [128].
A systematic review and meta-analyses of studies of exogenous surfactant treatment in ARDS between 1996 and 2005 concluded that exogenous surfactant may improve oxygenation but did not improve mortality. Hence, the authors were of the opinion that exogenous surfactant should be considered an effective adjunctive therapy in ARDS [129]. However, a recent meta-analysis of 11 randomized controlled trials, conducted between 1994 and 2015, found a lack of benefit with regards to mortality and no improved oxygenation in adult patients with ARDS who were treated with an exogenous surfactant when compared to controls [130]. The interpretation of these disappointing results was however hampered by several confounders such as inhomogeneity (patient phenotype, co-morbid conditions) of enrolled populations, diverse causes of ALI/ARDS, timing of initiating treatment, administration of a variety of exogenous surfactants with varying composition and viscosity, different doses and volumes, issues with pre-dosing drug preparation and method of installation(slow liquid bolus, positional tilting, aerosolisation), varying durations of treatment, inexperience of units with the drug and a lack of lung volume recruitment manoeuvres following treatment. Serious adverse events, such as hypotension, hypoxia and bradycardia also occurred (Table 2). At least 8 trials included sepsis-related ALI/ARDS and only two trials focussed on ‘direct’ ARDS [131-133]. The one study evaluating a recombinant surfactant protein C–based surfactant was prematurely terminated because of a lack of improvement in oxygenation, coupled with the results of in vitro tests which revealed that the administered suspension may have had insufficient surface activity to achieve clinical benefit [132]. The calf lung surfactant study could be considered a late rescue trial, since patients were randomized within 48 hours of intubation. This trial reported no difference in outcome between surfactant and placebo groups, and recorded transient hypoxia and hypotension associated with surfactant instillation [132]. Interestingly, patients diagnosed with H1N1 related ALI/ARDS, exhibited a low mortality (19%) compared to the Patient post hoc analyses suggest that the pathology with direct form of ALI/ARDS may achieve survival benefit from intratracheal surfactant. However, Wilson and colleagues failed to confirm an oxygenation or survival benefit when surfactant was used in ARDS [133].
Table 2. Summary of surfactant trials performed in direct and indirect ALI/ARDS in adults of between 2000 and 2020
Study (Year) Reference |
Surfactant |
Dosage
|
Delivery method or technique |
Trial / Population |
Possible confounders |
Findings |
SAE |
Spragg (2003)
[144] |
rSP-C (Venticute) |
1 mg of rSP-C per 50 mg PL/kg BW) or 0.5 mg of rSP-C per 50 mg PL/kg BW, versus controls.
X 4 over 24 hours |
Liquid bolus Tilted Catheter trough ET |
Pilot study; RCT; mixed (sepsis in ~ 30%) |
Disconnection; Tilting;
No SP-B; ~ 30 hours
diagnosis to treatment.
Dosage and duration of treatment |
No sign benefits. No improved ST lowering function. Decreased IL-6 levels in surfactant group |
Nil |
Spragg (2004)
[135] |
rSP-C
(Venticute) |
Up to 3 additional doses at 4-hour intervals within a period of 24 hrs |
Liquid slow bolus |
Multi-centre RCT; Various causes of ALI/ARDS |
Tilting; Several minutes between positional changes |
Improved oxygenation, but no improvement in survival |
4% surfactant group: hypoxia, hypotension, bradycardia |
Kesecioglu (2009)
[136] |
HL 10
Freeze-dried natural porcine
1-2% SP-B, C |
100 mg vials
3 g dispersed in 60 ml warm Saline: 50 mg/ml after dispersion.
Dosage: max cum dose 600 mg (200 mg/kg/dose), divided in 3 doses (0, 12, 36 hours) |
Large liquid bolus; 2 aliquots / tilting |
Multi-centre RCT; surfactant vs controls / ‘mixed’ ALI/ARDS |
Diagnosis to treatment delay (< 8 hours); Dispersion in warm saline required.
Tilting; Disconnection from vent during instillation |
No improved outcome
Post hoc: sign decrease in PaO2/FiO2 ratio up to 4 hours after surfactant |
Transient hypoxaemia; hypotension |
Lu (2010)
[206] |
HL 10 - as above |
X 3 doses
HL 10 delivered as above |
Large liquid bolus; recruitment manoeuvre |
RCT; surfactant vs controls. N=20; MV for ALI/ARDS (30% extra pulmonary sepsis) |
As above |
Surfactant increased lung reaeration, but failed to improve oxygenation / gas exchange |
NR |
Spragg (2011)
[132] |
rSP-C
(Venticute) |
Dry powder reconstituted; 100 mg/kg; Usual care plus up to 8 doses of rSP-C surfactant over 96 hours |
Liquid bolus |
Multi-centre RCT:
Surfactant vs controls ‘Direct’ ALI/ARDS (pneumonia, aspiration) |
Resuspension, shearing step introduced with this study |
No clinical benefit to patients with direct ALI/ARDS |
Transient hypoxia or airway obstruction |
Willson (2015)
[133] |
Calf lung wash extract
1mg% SP-B, C |
60 mg/ml PL
30 mg/cm
12-hrly X 3 |
Divided liquid bolus /Tilting |
Multi-centre RCT / Direct ALI/ARDS.
adults 18-85 years |
Tilting; Large liquid bolus.
Diagnosis to treatment delay (< 48hr); Vent. settings unchanged |
No improved oxygenation Trial discontinued of H1N1 cases, only 19% died. |
Transient hypoxaemia
Hypotension |
Pang (2017)
[88] |
Porcine-derived (Curosurf®) |
100 mg/kg, daily |
Liquid bolus after brief intubation, followed by extubation and NIV |
Mixed ALI/ARDS |
NR |
Improved oxygenation, duration in O2, less time on MV, fewer complication (2%) |
Infection (2%) |
PL: phospholipid; SAE: serious adverse event; Vent: ventilator; init: initial; MV: mechanical ventilation; max: maximum; cum: cumulative; sign: significant; Tilting: refers to turning patient on one side; NR: not recorded; ET: Endotracheal tube; ST: surface tension; O2: oxygen.
Post-hoc analyses of clinical trials of recombinant surfactant protein-C (rSP-C) therapy in ALI/ARDS confirmed improved oxygenation across the gr[134,135]oups studied and a significant improvement in survival in patients with pneumonia or aspiration induced ARDS [134]. A 2006 overview on the potential role for surfactant suggested that this approach warranted further study [128].
On a pathophysiological basis, there was an expectation that exogenous surfactant may assist in the management of the ALI/ARDS patient. However, it was somewhat optimistic to expect that correcting a single portion of the pathophysiology, would translate into improved survival outcome. A more correct approach would have been the evaluation of compliance and oxygenation before and after surfactant. As noted by Kesecioglu et al. [136], neonatal or paediatric patients who suffers from acute respiratory distress, often die as a result of respiratory failure whereas adults with ALI/ARDS mostly die as a result of multi-organ and or right heart failure. Hence there was an expectation that surfactant will be more effective treatment in paediatric patients compared to adults when survival is the measured outcome. The authors reported on the mortality of an international, multi-centre, randomized trial of adult ALI/ARDS, resulting from a wide variation of causes. A bolus of natural porcine surfactant HL 10 treated group, compared to the usual care group, started to show a worse outcome trajectory approximately 3 weeks after beginning of the trial. The reason for the different patterns was unclear, but the authors speculated it could be related to factors other than the administered surfactant or underlying ARDS [136].
Animal-derived surfactant preparations, containing various amounts of specific hydrophobic proteins SP-B and C, are clearly effective in preventing and treating nRDS in premature infants and infants diagnosed with meconium aspiration. Exogenous surfactant replacement has been successful in numerous trials involving neonatal RDS, several smaller studies of neonatal ALI/ARDS caused by meconium aspiration and in young infants with severe viral-induced bronchiolitis [137-143]. In contrast, clinical trials in adults with ALI/ARDS produced mixed and mostly disappointing results [91,141,144].
Perhaps one of the reasons for lower activity of surfactants in treatment of ALI/ARDS is the inactivation of the instilled drug. Researchers therefore shifted focus to the re-engineering of a synthetic surfactant preparation uniquely able to resist the negative effects on surfactant associated with leaked fibrinogen, haemoglobin and albumin into the alveoli and acute inflammation and oxidative stress [145-148]. There is also limited evidence demonstrating that a surfactant with synthetic, inhibition-resistant and anti-inflammatory characteristics will perform better than products which does not have these characteristics. The mammalian-derived formulations being an example of the latter [145-148]. One such synthetic preparation (CHF 5633), containing DPPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), surfactant protein B and C analogues, showed significant benefit when compared to animal derived surfactant (Curosurf®) in an in vivo model of surfactant inactivation in premature lambs [146].
The synthetic surfactant (Synsurf®), evaluated by van Zyl et al. [147,148] consists of DPPC and PG and a poly-L-lysine–poly-L-glutamic acid construct. It is suggested that this formulation mimics the SP-C /B structural and functional properties and therefore may well promote its anti-inflammatory function and provide improved resistance to inhibition. This expectation was recently confirmed in a study by Van Rensburg et al. [149]. The authors employed proteomics to compare the potential immune modulatory properties of two natural surfactants with the synthetic surfactant Synsurfâ on the pro-inflammatory cytokines TNF-α, IL-1β, IL-6, the oxidative burst and protein expression in lipopolysaccharide stimulated alveolar macrophages. The study demonstrated that surfactant cause a decrease in secretion of the pro-inflammatory cytokines, TNF-α, IL-1β, and IL-6 in a dose dependent manner. Moreover, the surfactants inhibited the oxidative burst of stimulated alveolar macrophages by decreasing reactive oxygen species (ROS) production. In addition, the synthetic surfactant Synsurfâ, up regulated Peroxiredoxin-1 (Prx1), a family of antioxidant proteins which protects the cell from metabolically produced ROS that usually trigger toxic mechanisms within cells. The authors hypothesised that this up regulation of Prx1 ultimately contributed to the modulation of redox-sensitive cellular behaviours whenever the signal for oxidative damage is exacerbated or continues. They also speculated that Prx1 perhaps inhibit NO production by suppressing the ROS/NF-κB/iNOS (NOS2) signalling pathway. The latter may ultimately aid in the decrease in cytokine production associated with Synsurfâ-exposed stimulated macrophages via blocking the induction of NF-κB transcription. In addition, upregulated Coiled-coil domain protein 22 (CCD22), found in the Synsurfâ exposed alveolar macrophages, also contributed to the proteasomal degradation of NF-kB, which will further add to the modulation of the JAK/STAT pathway and eventually have an effect on the pro-inflammatory cytokine production. The same study also found that Trx1, an oxidative stress-limiting protein, was up regulated within the Synsurfâ group, this being one of the important cellular antioxidants with anti-inflammatory and anti-apoptotic properties [149]. Differentiation of macrophage into M2 phenotypes was promoted by reducing inflammatory M1 macrophages which leads to a dose dependant decrease in TNFa and IL 1b expression. All of the preceding, taken together would suggest that Synsurfâ,a versatile mediator of inflammation, could be a possible candidate for the treatment of pulmonary inflammatory disorders besides acting as a surface tension stabilizer agent in alveoli [149].
Recently, Ahlstrom et al. studied acute respiratory distress syndrome (ARDS) in the adult rabbit model [150]. Surfactant, based on a recombinant surfactant protein C analogue (rSP-C33Leu), was instilled and its effects on lung function and inflammation compared to a commercial surfactant (Curosurf®) and air as control. SP-C33Leu contains a poly-leucyl transmembrane α-helix, a positively charged residue in the N-terminal part of the helix to avoid oligomerization and, in addition, a methionine residue was replaced with leucine to avoid in advertent oxidation. The animals were treated with two intratracheal boluses of 2.5 ml/kg of 2% rSP-C33Leu in DPPC/egg PC/POPG, 50:40:10 or poractant alfa (Curosurf®). Both surfactants contain 80 mg phospholipids/ml. Animals were subsequently ventilated (VT 8–9 m/kg body weight) for an additional 3 hours and lung function parameters recorded. Histology of the lungs, degree of lung oedema and levels of the cytokines TNFα IL-6 and IL-8 in lung homogenates was evaluated. Both surfactant preparations improved lung function, reduced inflammation scores production of pro-inflammatory cytokines and formation of lung oedema. Poractant alfa improved compliance at 1 h, PaO2/FiO2 ratio and PaO2 at 1.5 h compared to rSP-C33Leu surfactant. The authors concluded that results indicated that treatment of experimental ARDS with synthetic lung surfactant, based on rSP-C33 Leu, improved lung function and attenuates inflammation.
Phospholipase A2 and C show increased activity in the bronchoalveolar lavages of patients with ARDS. It is therefore reasoned that a surfactant supplemented with DL-alpha-Di-O-hexadecylphosphonocholine [(R)(S)-DEPN-8], which is not a substrate for PLA1, A2 or D, but a substrate for PLC, could be incorporated as component to a surfactant formulation for treatment of ARDS [151]. (R)(S)-DEPN-8 undergoes less PLC-catalysing as compared to DPPC. Synthetic surfactants containing DEPN-8 or other phospholipase resistant lipids plus active SP-B peptides, in theory, may have particular utility in treating ALI/ARDS. New synthetic surfactants can also potentially be bioengineered to include novel peptide components incorporating the most active regions of other human surfactant apoproteins in combination with SP-B peptides and lipids [146].
In ALI/ARDS Schmidt et al. [121] found a significant correlation between the PaO2/FiO2 ratio and minimum surface tension of the alveoli when SP-C and DPPC were administered to intubated patients (average time from diagnosis of ARDS to initial BAL was 21±2 hours). Although there was an initial improvement in surfactant function over time, compositional surfactant abnormalities and poorer surface tension reducing ability persisted for 7-9 days following ventilation. The authors correctly questioned the use of a single administration of surfactant given that the lung abnormalities lasted for a considerable time.
When temporal changes in pulmonary surfactant function and composition in ALI/ARDS, due to pneumonia or aspiration, are considered, exogenous surfactant may have to be administered according to an ‘early rescue’ strategy and perhaps continued thereafter. Considering such an approach would ensure early and continuous treatment of patients at risk for developing serious ARDS. However this approach may well result in the unnecessary treatment of patients and the concept needs further examination and prospective evaluation. In newborn infants, with secondary surfactant deficiency or dysfunction related to meconium aspiration or pneumonia, two RCTs reported benefits of surfactant replacement therapy. One studied infants requiring 100% oxygen with an oxygenation index (OI= product of mean airway pressure (cm H2O)), FiO2 and 100/PaO2 (mm Hg) greater than 15 [96], and the other studied infants requiring more than 50% oxygen with an arterial/alveolar O2 tension ratio of less than 0.22 [95]. Point being that in both these studies the advantage of treatment with surfactant was evident if used early in the disease.
However, in more recent adult trials, there was a delay between onset of mechanical ventilation and initiation of a single dose treatment with surfactant. In one trial, porcine-derived surfactant (HL 10) was administered at a median of 35.4 hours (range: 2.4 - 74 hours) [136]. In patients (12 and 85 years of age) with severe direct lung injury due to pneumonia or aspiration, a single dose exogenous recombinant surfactant protein C (rSP-C)-based surfactant was administered when the PaO2 /FiO2 ratio was below 170. In the surfactant treatment group this level was reached around 100 hours after being hospitalized and 37 hours from intubation [127]. None of these studies showed a significant functional improvement or better outcome. In patients diagnosed with ARDS after cardiac surgery, bronchoscope directed bovine derived surfactant (a twice daily dose) was administered ‘within 24h’ of the start of ARDS (oxygenation index below 200 mm Hg) [152]. This study showed improved oxygenation and survival when compared to controls. In the calfactant ARDS (CARDS) trial, surfactant was administered within 48 hours of initiation of mechanical ventilation if the SpO2/FiO2 was less than 250 (when SpO2 < 97%) [133]. The results were disappointing inasmuch as it did not show an improvement in oxygenation or survival.
With regards to the question of what is implied by “early” treatment, the Berlin criteria is probably not of value as it defines the condition (for ARDS) already being present. However, if the “mild” form of ARDS or even ALI is viewed as perhaps being early, it could signal the time to start continuous exogenous surfactant treatment. Although not the purpose of this overview, the use of biologic predictors, in addition to clinical markers (P/F ratios, chest X rays CT scan, level of PEEP, etc.), may have the capacity to improve prediction of outcome and risk stratification. Some studies suggest that plasma biomarkers, such as interleukin-8 (IL-8), tumor necrosis factor alpha (TNFα), surfactant protein-D (SPD), and mitochondrial DNA, advanced glycation end products (sRAGE) as a marker of lung, epithelial injury, plasma levels of tumor necrosis factor receptor-1 (TNFR1), IL-6, and plasminogen activator inhibitor-1 (PAI1) as markers of a hyperinflammatory ARDS sub-phenotype, may assist in identifying patients at risk of developing severe disease [153]. Whether these markers would assist to timeously identify candidates with mild ARDS for early rescue surfactant treatment, is not known and, in our view, is worth pursuing as a concept.
The median (IQR) duration of surfactant treatment in adult patients with ARDS (11 randomized trials) conducted between 1994 and 2010, was 36 (24-96) hours [130]. Two trials extended treatment up to 5 days. In the very early studies, aerosolized, synthetic protein-free surfactant (Exosurf®), was administered (without any effect) on 10 and 28-30-day mortality [130]. Gregory et al. [153] studied 4 different groups of patients, with diverse causes for their ARDS. Treatment was given within 48 hours of diagnosis. Patients were treated with bovine surfactant for up to 96 hours. Significantly lower FiO2 at 120 hours and a significant decreased risk of death for the 100 mg/kg x 4 doses groups were reported when data was compared to that obtained from standard therapy without surfactant.
Spragg et al. found that the frequency of surfactant administration is likely to be an important variable after their study with recombinant surfactant protein-C. The increased fractional content of PG and increased levels of SP-C detectable in BAL obtained at 48 hours, but not at 120 hours after the initial administration, lead them to conclude that it may be beneficial to administer surfactant over periods longer than 24 hours, perhaps 120 hours or more [144]. Considering the aforesaid, and the findings of Schmidt et al. [121], it seems reasonable to speculate that the duration of treatment with exogenous surfactant, i.e. multiple dosing (in combination with less injurious lung ventilation strategies), must be extended beyond the first 5 days of the disease in order to (perhaps) effectively influence surfactant composition and alveolar surface tension behaviour.
The optimal dose, the interval between diagnosis and treatment, number of doses, duration of treatment with therapeutic surfactant for adult ALI/ARDS, has not yet been clarified. In a group with diverse causes underlying ARDS, Gregory et al. [153] found that the volume of bovine-derived surfactant (4 ml/kg, eight times to a 70 kg patient) instilled into trachea, was generally well tolerated. The most common side effect was reflux of the surfactant into the endotracheal tube, which occurred most often in the 50 mg x 8 group and least often in the 100 mg x 4 group. In this study, the instilled volumes of surfactant, at 100 mg/kg (4 ml/kg), to a 70-kg patient, would have amounted to a single dose of 280 ml of liquid surfactant. Subsequently, a controlled trial with calfactant in adult patients, with direct ARDS, reported no clinical benefits, including no improvement in oxygenation [107]. In this unsuccessful trial, surfactant was administered as a low volume-concentrated dose (60 mg/ml, approximately 1.3 ml/kg) [154]. This volumetric dose was approximately 32% of the volume used in the Gregory et al. trial [153]. Another unsuccessful controlled trial of surfactant therapy in adults with ARDS by Spragg et al. [132] used concentrated low-volume doses (1 ml/kg) of instilled recombinant surfactant protein C–based surfactant (Venticute).
Considering the disappointing trials with surfactant in adults and the much larger conducting airway surface of the adult lung, the question must be asked if the instilled surfactant mixtures, used in these trials, were of sufficient volumes. Perhaps the surfactant failed to reach the majority alveoli/acini [154,155]. 3D modelling of the airway transport of intratracheal surfactant revealed that the instilled volumetric dose is a critical independent parameter to achieve sufficient distribution of the drug [155]. This study found that, in contrast to the neonatal lung, the adult lung did not behave as a ‘well-mixed’ compartment and consequently the lower volumetric doses of surfactant only coated the airways, rather than reaching the alveoli. Furthermore, it was speculated that lower volume - higher surfactant concentration, may have increased viscosity at low shear rates, further adversely affecting distal delivery.
Grotberg et al. [154] suggested that coating of the conducting airways significantly reduces the amount of instilled surfactant arriving at the alveoli (labelled as the “coating cost”). This suggests that the instilled dose volumes must be well above the coating cost in order to ensure delivery to the alveoli. Furthermore, the delivery efficiency of surfactant may decrease if the ventilator airflow rate increases.
Bronchoscopy instilled bovine surfactant, at much larger volumetric-concentrated doses, to 27 cases of septic patients with early ARDS, resulted in pronounced and significant pre to post treatment improvements in gas exchange within 12 hours (increased PaO2/FiO2 ratio by approximately 93%) as well as improvement in the biochemical composition and surface tension lowering properties of surfactant isolates [156,157]. This trial instilled surfactant at an initial dose of 300 mg/kg body weight (mean total volume 378 mL or at approximately 5 - 6 ml/kg ~60 - 66 mg/ml PLs) to a 70 kg patient. Portions of the total dose were delivered to each segment of the lungs via flexible bronchoscope within approximately 45 min. In 7 of the patients, in whom gas exchange again deteriorated, a second surfactant dose of 200 mg/kg (~3 ml/kg) was administered 18-24 hours after the first application, again improving arterial oxygenation within 1 hour by approximately 30%. The authors reported that the total amount of administered surfactant material (300 - 500 mg/kg) surpassed the endogenous surfactant pool. When considering the higher volumetric, concentrated or less concentrated, doses of surfactant used in selective trials and the 3D modelling, it is predicted that the adult patient may require a higher volumetric dose to coat the large conducting airways and reach the alveoli [154]. One can speculate that if liquid boluses of mammalian-derived surfactant are administered, the volumetric dosage should not be less than 60 - 66 mg/ml PLs, i.e. no less than 3 - 5 ml/kg or 200 - 500 mg/kg body weight [154,155,157-160]. From the above data it seems that if the tracheal installation approach is followed, the practitioner must figure in the coating cost i.e. use larger volumes. If the surfactant is going to be delivered by fibre optic bronchoscope into the lobar or segmental bronchi, smaller doses may well suffice.
In the study where surfactant (bovine (calf lung) extract, 3 - 5 ml/kg (approximately 70 kg patients) was delivered via bronchoscopy into patients with severe ARDS, it was found that in approximately 26% of cases, the initial improvement in gas exchange was lost over time and retreatment with a second dose of surfactant was necessary within 18 - 24 hours. For healthy adult rabbits, the alveolar turnover time of PC has been reported to range between 5-10 ml/hour [17] and after intratracheal injection of labelled surfactant material, the overall loss from the airspaces and the lung reached 90% and 70%, respectively, after 24 hours [161]. The difficulty is to transpose the much faster rate of small animal metabolism to adult humans but it seems reasonable to assume that most of the applied surfactant material was cleared from the lung within the first day after treatment. It can perhaps be speculated that some of this material may re-enter the alveolar compartment due to recycling via type-II cells.
This experimental, and some of the clinical data, would suggest that the correct and repeated dosing may well be of value in adult ALI/ARDS. Not doing so could perhaps explain some of the reported unsuccessful results to date.
Studies on bolus liquid surfactant administration, as used in infants and children, have not shown significant adverse long-term effects. Liquid bolus surfactant instillation of intubated and ventilated adult cases with ALI/ARDS has been examined in trials. Transient hypoxia and some hemodynamic instability (hypotension) were reported at the time of intratracheal or bronchoscopic instillation of surfactant in adults [85].
The question is whether (repeated) bronchoscopically directed surfactant instillation is perhaps the preferred method of surfactant installation. Flexible bronchoscopically-directed instillation of surfactant allows deposition of the drug in close approximation of the alveoli. If this approach is combined with lung recruitment manoeuvres during and immediately after surfactant delivery, it may well lead to more homogeneous distribution within the lungs and result in a superior clinical response. The recruitment of alveoli can be achieved by intermittent sustained inspiratory positive airway pressure hold and the recruited alveolar volume can be maintained by proper PEEP titration. Although appropriate PEEP will also recruit alveoli over time, it may take too long given that the aim is to be able to deliver the surfactant into “open” alveoli. The surfactant will, in theory, add alveolar stability over and above the mechanical effect of PEEP [162-164].
There seems to be general support for the methods of surfactant delivery via a bronchoscope [165]. There are 13 reports describing bronchoscopic administration of surfactant to 88 human patients with ARDS (Table 3). The majority of these unfortunately were uncontrolled case series. The studies reported either partial or complete improvements in atelectasis, gas exchange or / and restoration of surfactant properties, lower MV settings or successful termination of mechanical ventilation. Where outcome was reported, 80% of cases survived. The study by Gunther et al. [156] overshadows the smaller case series reports and showed that large doses of natural bovine surfactant almost fully restored biochemical properties and resulted in much improved biophysical surfactant function.
Table 3. Human studies of bronchoscopically instilled exogenous surfactant
First author (year) Reference |
Study design |
Number of patients (n) |
Patient type |
Disease |
Process and Surfactant |
Outcomes |
Satoh (1998) [207] |
Controlled |
6 |
Adult |
Burn patients with ARDS |
Prior suctioning of airways
Alveofact (50-100 mg/kg BW
Repeated 24-hrly if needed |
All survived.
Temporary improved oxygenation and compliance
Repeated dosages required |
Spragg (1994) [87] |
Crossover |
6 |
Adult |
ARDS |
Following single lobe BAL, porcine-derived (4 g in 50 ml; 50-60 mg/kg BW) in aliquots to each lobar bronchi |
Transient improved gas exchange, without changes in CXR or markers of lung inflammation. Four (66%) survived |
Walmrath (1996) [157] |
Uncontrolled |
10 |
Adult |
ARDS and sepsis
MOF: 90% |
Prior single segment lavage;
Alveofact® (300 mg/kg; 313 ml Saline);
Segmental instillation;
Duration: 50±12min |
Improved oxygenation decreased shunt flow.
Retreatment in 50% after 18-24 hr
50% survived. |
Hoheisel (1997) [208] |
Case report |
1 |
Adult |
Sepsis/ARDS, persistent atelectasis |
Independent ventilation of both lungs; selective Alveofact® (1 gram) to left lung |
Improved oxygenation and imaging |
Staudinger (1997) [209] |
Case report |
1 |
Adult |
ARDS (near-drowning) |
Curosurf™ (50 mg/kg) to each lung |
Transient improved PaCO2, FiO2 and shunt fraction.
Repeat treatment (X6; 300 mg/kg total) required.
Patient died due to sepsis |
Violi (1997) [210] |
Case series |
3 |
Paediatrics (n=1) and Adult (n=3) |
ARDS |
Segmental instillation of Survanta® (4 ml/kg).
Procedure duration: 20 min |
No complications |
Pallua (1998) [211] |
Descriptive |
4 |
Adult |
Burn patients with ARDS |
After limits of MV reached, saline lavage, followed by intrabronchial instilled Alveofact® (50-152 mg/kg) |
Temporary improved gas exchange and compliance and CXR clearance.
One required retreatment (X2).
All survived |
Wiswell (1999) [212] |
Uncontrolled |
12 |
Adult |
Variety cause-ARDS |
Bronchopulmonary segmental lavage (19 segments) with dilute synthetic surfactant (KL4), followed by suctioning.
Groups received 82-114 mg/kg surfactant, diluted in 1710 ml.
Procedure duration: averaged 1½ hrs.
Maintenance of PEEP≥24hrs |
In the 96 hrs after treatment, improved oxygenation and PEEP levels
No serious adverse events.
Nine (75%) survived. |
Nakamura (2001) [165] |
Case report |
1 |
Paediatric |
ARDS (Herpes virus 6) |
Survanta®, 140 mg/kg (64 ml), instilled into 4 main bronchi; duration ~ 30 min |
Improved oxygenation and lower MV settings.
Patient died |
Gunther (2002) [156] |
Controlled |
27 |
Adult |
ARDS and septic shock |
Segmental instillation (Alveofact®, 300-500 mg/kg BW; ~378 ml saline). Procedure duration: 45±11min.
Retreatment allowed after 18-24 hrs |
Large amount of surfactant restored PL profile, LA fraction and SP-B and SP-C content and improved ST properties.
Improved PaO2/FiO2 in majority; retreatment in 7% |
Tsangaris (2007) [164] |
Randomized |
16 |
Adult |
Multiple trauma, lung contusion and ARDS |
Alveofact® instilled into involved lung areas; each segmental bronchus received (200/19) mg/kg BW. |
Compared to controls, improved oxygenation and compliance.
Better response with recruitment manoeuvre.
All survived |
Krause (2008) [213] |
Uncontrolled |
5 |
Paediatric |
Persistent lobar atelectasis (ARDS, CF, CDH, hemi-Fontan circulation) |
Suctioning, followed by diluted (5-10 mg/ml) Curosurf™ (120-240 mg) into affected segments |
Improved oxygenation, respiratory rate and partial or complete resolution of atelectasis.
All extubated < 24hrs |
Krause (2014) [214] |
Case series |
4 |
Paediatric |
ARDS due to RSV-pneumonia |
Suctioning followed by segmental BAL with diluted (20 ml) Curosurf™ (6 mg/ml) and rhDNase (1.25 mg).
Procedure duration: 25-40min |
Resolution of atelectasis < 8 hrs in 65%.
Restoration of gas exchange and compliance.
All survived |
Alveofact®: Bovine-derived surfactant (Thomae, Biberach/Germany); Curosurf™: Porcine-derived surfactant (Chiesi Pharmaceutici, Parma, Italy); KL4; Discovery Laboratories, Doylestown, PA); Survantaâ: Bovine-derived surfactant (Ross, Columbus, OH); MOF: multi-organ failure; CXR: Chest x-ray; BAL: Broncho-alveolar lavage; BW: Body weight; MV: Mechanical ventilation; PL: Phospholipid; LA: Large aggregate; ST: Surface tension; CF: Cystic fibrosis; CDH: Congenital diaphragmatic hernia; RSV: Respiratory syncytial virus.
Bronchoscopy directed surfactant treatment however, requires skilled medical staff and is relatively labour intensive. The procedure is generally well-tolerated and performed within approximately 1.5 hours. The studies report minimal or transient adverse events. It appears that higher doses may have to be used to overcome inhibitory substances in the alveolar space, as was shown in animal models [166]. The more recent adult calfactant, randomized, controlled trial reported a lack of improved oxygenation or longer-term benefits relative to placebo [133]. Surfactant instillation was associated with significant but transient adverse effects, primarily hypoxia and hypotension, leading the investigators to state that further studies of exogenous surfactant administration should consider using recruitment manoeuvres during instillation and that exogenous surfactant cannot be recommended for routine clinical use in ARDS.
If effective surfactant delivery via aerosolization can be perfected, it may avoid the transient hypoxia, hypertension, and partial endotracheal tube obstruction associated with bronchoscopy directed surfactant instillation.
Nebulisers convert solutions and suspensions into small droplets. The advantage of using nebulisers includes their ability to aerosolise high doses of drugs that are not available with dry powder inhalers (DPIs) or pressurised metered dose inhalers (pMDIs). Efficiency of drugs delivered to the lung is significantly affected by particle size. Large particles (5 - 10 µm) tend to deposit mainly in the upper airway (trachea and primary bronchus) by means of impaction associated with turbulent and high air flow velocity commonly found in with aerosol delivery. This limits the amount of drugs that can be delivered to the alveoli. Small particles (< 2 µm) deposit mainly in the alveolar region, whereas the particle size range 1 - 5 µm deposit primarily in the bronchus and bronchioles [167]. Particle smaller than 0.5 mm is expected to be expelled upon exhalation. The latest guidelines for aerosol therapies recommend slow inhalations (< 30 L/min flow) and are based on evidence gathered from pMDIs studies [168]. Most nebulizers available produce a mass median aerosol diameter of 6 µm or less, suggesting that the devices will allow for delivering aerosolized drugs to the lung segments according to air flow distribution [167]. In the periphery of the lung, airflow rate is reduced and particles deposit predominantly by sedimentation, with gravity causing them to “rain out” and deposit itself on the available surface [167,168].
Newer-generation vibrating mesh nebulisers have been designed specifically for use during mechanical ventilation and have now also become the first choice for nebulized pharmaceutical drug delivery [168,169]. Vibrating mesh devices seem to have advantages over other nebuliser systems and are being developed for other uses, such as the delivery of vaccines and nebulising liposomal formulations and proteins [168]. However, vibrating mesh devices may result in variable dose output related to the viscosity of surfactant. Studies with nebulized surfactants have reported clogging of the pores of the vibrating membrane of the nebulizer, requiring surfactant dilution before nebulization [170].
Aerosol delivery would be ideal in terms of ensuring uniform alveolar distribution of lung surfactants. This method can avoid potential complications associated with repositioning patients from side to side during bolus instillation of lung surfactants, particularly in a patient population with direct and homogeneous lung injury. As mentioned earlier, the greatest advantage of aerosol delivery is that an aerosol could be delivered with, for instance, a CPAP mask or in other forms of non-invasive breathing support.
However, aerosol delivery has limitations. It provides surfactant only to areas of the lung which are actively participating in ventilation. Hence in a heterogeneous lung injury (such as the histology of ALI/ARDS shows, see Figure 2), the areas of the lung that do not actively participate in ventilation, are the alveoli which probably are most deprived of surfactant and therefore, in theory, should benefit most from the surfactant delivery [85,146,171,172]. Secondly, aerosol delivery of drugs is, in general, mostly suitable for inexpensive medications. The relative amount of surfactant that will actually be deposited in the lung after aerosolisation is likely in the 5 – 15% range [131]. Thus, to provide a clinically effective dose, much more material is required compared to bolus instillation. This cost factor would be less of concern if the drugs were cheap [173].
In the early Exosurf® (synthetic protein-free surfactant) trial, surfactant was delivered by continuous aerosolisation. Only 4.5% of aerosolized radiolabelled surfactant reached the alveoli. Thus, < 5 µg of 102 µg of aerosolized DPPC/kg/day reached the alveoli in this study [131].
In a sheep model with non-uniform lung injury, induced with HCl, Lewis et al. [174] demonstrated superiority in both oxygenation and ventilatory parameters in animals treated with aerosolized surfactant administration by low-flow nebulizer when compared to surfactant (Survanta®) instilled via the trachea. Very little (8%) of the aerosolized surfactant was deposited in the more severely affected portions of the lung, presumably reflecting poor ventilation of these areas. The majority of the surfactant was recovered from the rest of the lung.
Recently, Bianco et al. [170] investigated the feasibility of delivering nebulized, undiluted porcine-derived surfactant (poractant alfa) in vitro and in vivo (spontaneously breathing rabbits with respiratory distress) with an adapted, neonate tailored aerosol delivery strategy (aerosols generated by a customized eFlow-Neos vibrating-membrane nebulizer system). In this study, nebulized surfactant was delivered at doses of 200 mg/kg and 400 mg/kg. The effect on respiratory mechanics and arterial oxygenation in the 400 mg/kg group were similar as for the 200 mg/kg group and these were significantly better than that obtained in the control group (no surfactant).
Synthetic surfactants, 3% Super Mini-B peptide (SMB surfactant), a highly surface active SP-B clone , and a combination of 1.5% SMB and 1.5% of the SP-C mimic SP-Css ion-lock 1 (BC surfactant), with a synthetic lipid mixture (DPPC:POPC:POPG 5:3:2 weight ratio) were administered via a vibrating membrane nebulizer to saline-lavaged surfactant-deficient rabbits who were supported by either nasal CPAP or mechanical ventilation (MV) [175]. Particle size of the surfactant aerosol was within the 1-3 µm range and surfactant activity was not affected by aerosolisation. At a dose equivalent to clinical surfactant therapy in premature infants (100 mg/kg), aerosol delivery of both synthetic surfactant preparations led to a rapid and clinically relevant improvement in oxygenation and lung compliance in the rabbits, more so in rabbits supported by mechanical ventilation. Lung delivery of surfactant aerosol via nCPAP was less efficient than intratracheal aerosol delivery because of the retention of nebulized surfactant by the circuit and loss during its passage through the nasopharynx and upper airways. The authors suggested that increasing the lung delivery dose can be achieved by using a higher surfactant dose, and/or longer delivery times of surfactant aerosol or by decreasing the viscosity by dilution with saline or water. These suggestions require further testing.
In an in vitro study, the biophysical properties of particles of the synthetic surfactant Synsurfâ, was investigated with the use of an AeronebâPro vibrating mesh nebuliser. Particle generated during nebulisation of different Synsurfâ formulations was below the recommended range of 1000 d.nm-3000 d.nm, suggestive for optimal peripheral lung deposition (Z-average particle size expressed as diameter in nanometers). Research again established that the biophysical properties such as the size and density, determined the deposition efficiency of the surfactant [176].
Studies on the biophysics of the respiratory cycle show that the reduction in surface tension (g) at any point on the air-liquid interface is proportional to surfactant monolayer density [100,177]. With the aid of computational models of alveolar forces and lung architecture, it was shown, with molecular dynamic simulation studies, that the microscale fluid-mechanical forces in the recruitment and de-recruitment of small airways and alveoli, involves complex interactions between surface tension forces, surfactant transport, fluid flow and the solid mechanics of the compliant airway. This complex interaction makes studying surfactant transport challenging, - more so with time-dependent flow and or with flexible airway walls.
Simulations of steady reopening in rigid [178] and compliant [179,180] domains suggest that the slow adsorption rate of pulmonary surfactant may, in some cases, increase the pressure required to reopen an airway when compared to constant surface tension at the equilibrium value. This occurs because variations in surface tension along the interface result in tangential Marangoni stresses which rigidify the interface [181]. In order to side-step the limitations of slow surfactant adsorption, Smith et al. modelled the occluded airway as a liquid-filled rigid tube with the fluid phase displaced by a finger of air. With the aid of computational analysis techniques applied to surfactant free velocity fields, they proposed adding a sinusoidal component to the driving flow (technique and technology of administering surfactant) so that the bubble retracts periodically instead of it progressing steadily forward [177,182-184]. This in vitro experimental approach studied surfactant distribution from the bulk phase across the air-liquid interface. It demonstrated a 50% improvement in surfactant function [185]. These findings suggest that ventilation waveforms could be designed to maximize the efficacy of pulmonary surfactant and thereby minimise the risk of VILI [186].
This concept originated from neonatal respiratory medicine and is referred to as minimal invasive surfactant treatment [187].
In a small case controlled study, probably the first in adults with diverse causes of ALI/ARDS, patients were treated according to a strategy of ‘less invasive surfactant administration’ (LISA) utilizing a technique of surfactant instillation similar to the INSURE (INtubation-SURfactant-Extubation) strategy, already popularized in premature infants with nRDS [187]. Patients in the control group were treated with conventional mechanical ventilation while those in the observation group were given porcine-derived surfactant (Curosurf®) as a daily dose of 100 mg/kg. Although it is not clearly stated, it appears that the authors intubated the surfactant group, administered surfactant, and then after a short period of ventilation (3-5 minutes), extubated the treatment group and continued with CPAP, very similar to the INSURE strategy. Compared to the control group, the treated group performed better in terms of gas exchange and time spent on mechanical ventilation.
Alternatively, a laryngeal mask, instead of an endotracheal tube, could be used for early surfactant treatment to spontaneously breathing patients during the early course of ALI/ARDS. Once administered and extubated, CPAP or HHFNO2 should be continued and hopefully again assist in avoiding invasive mechanical ventilation.
During hyper inflammation and acute respiratory distress syndrome, similar to patients with SARS-CoV and MERS-CoV, some patients with COVID-19 develop acute respiratory distress syndrome (ARDS) with characteristic pulmonary ground glass changes on imaging [188]. Evidence confirms that some of the severe COVID-19 patients have an elevated cytokine profile resembling the cytokine storm described in SARS and MERS [189]. The observations have been found consistent with the characteristics of the so called “primary cytokine” storm induced by viral infections which were mainly produced by alveolar macrophages, epithelial cells and endothelial cells. This appears to be different to the cytokine response observed in “secondary cytokine” storm induced by different subsets of activated T lymphocytes in late stage of viral infection or of a complication of T cell-engaging therapies. In line with these observations, Liao et al. [190] showed that monocyte-derived FCN1+ macrophages, but not FABP4+ alveolar macrophages which represent a predominant macrophage subset in BAL in patients with mild diseases, overwhelm the severely damaged lungs of patients with ARDS. These cells have a high inflammatory potential and are producers of large amounts of the chemokines implicated in cytokine storm.
Fu et al. [191] explored the possible mechanisms of the inflammatory response observed in COVID-19 pneumonia. Based on previous studies of SARS-CoV, they separated the inflammatory responses. In SARS-CoV-2 infection into primary and secondary responses Primary inflammatory responses occur early after viral infection, prior to the appearance of neutralizing antibodies (NAb). These responses are mainly driven by active viral replication, viral-mediated ACE2 downregulation and shedding and host antiviral responses. Secondary inflammatory responses begin with the generation of adaptive immunity and NAb. The virus-NAb complex can also trigger FcR-mediated inflammatory responses and acute lung injury.
The inflammatory process related to ALI/ARDS (in general) may be limited to the lung, with lower levels of inflammatory mediators in the systemic circulation [90,192]. However, compared to other viral causes of ALI/ARDS, there is evidence showing that a subgroup of patients with severe COVID-19, experience a cytokine ‘storm’, i.e. virally driven hyper inflammation [118]. A cytokine profile resembling secondary haemophagocytic lymphohistiocytosis (sHLH) is associated with COVID-19 disease severity, characterised by increased interleukin IL-2, IL-7, granulocyte colony stimulating factor, interferon-γ inducible protein 10, monocyte chemoattractant protein 1, macrophage inflammatory protein 1-α, and tumour necrosis factor-α [118]. Although corticosteroids were not routinely recommended during previous pandemics, it is now realized that immune suppression with steroids, selective cytokine blockade and Janus Kinase (JAK) inhibition, may alter the clinical course of COVID-19 infection [118,193].
Considering then that COVID-19 infects the type II pneumocytes (cells manufacturing surfactant) and what has been reported on the inflammation cascade in COVID-19 (summarized supra), we propose that exogenous surfactant treatment has the potential to alleviate the severity of the inflammation in the alveoli. This inflammation-suppressive effect of synthetic surfactants containing a peptide / protein , has now been convincingly been demonstrated in the studies by Ahlstrom et al. and Van Rensburg et al in the rabbit ARDS model and the LPS stimulated human alveolar macrophage [147,150].
Our own research compared the natural derived surfactants, Curosurf® and Liposurf®. For this purpose, we investigated cytokine production from BAL derived alveolar macrophages (AMs) treated with either Synsurf®, Curosurf® or Liposurf® [147]. Thirty children, aged 3 to 14 years, undergoing diagnostic bronchoscopy and bronchoalveolar lavage, were studied. Differential cytology, cytokine and chemokine measurements were performed on the fluid after exogenous surfactant exposure. The aim of the study was to investigate the potential anti-inflammatory effects of exogenous surfactants on the BAL fluid, specifically alveolar macrophages in healthy South African children. The results showed that alveolar macrophages were the predominant cellular population in normal children. Patients with inflammatory pneumonopathies had significantly more neutrophils in the BAL. Levels of inflammatory cytokines were significantly lower after exogenous surfactant exposure. Moreover, IL-10 and IL-12 cytokine secretion increased after exogenous surfactant exposure. We found a weak relationship between LPS stimulated release of IL-1β, IL-2, IL-6 and IL-8 and their spontaneous secretion during prolonged exposure to LPS in combination with surfactant. Synsurf® displayed an approximate two-fold decrease in IL-1β release (97.60 pg/ml) compared to control levels (176.45 pg/ml) whereas the natural surfactants displayed a minimal decrease. After 24 hours, TNF-α levels decreased within the cell supernatant of the surfactant treated AMs compared to controls. The Curosurf® (120.73 pg/ml) and Liposurf® (110.01 pg/ml) groups displayed an approximate four-fold decrease in TNF-α release compared to control levels (462 pg/ml) whereas, Synsurf® (56.87 pg/ml) displayed a much larger, eight-fold, inhibitory effect on stimulated TNF-α release. TNF-α and IL-1β release by macrophages are “acute response” cytokines that promote neutrophilic and eosinophilic inflammation. Although they are not directly chemo-attractive agents, they may directly or indirectly stimulate the upregulation of relevant secondary cytokines and cell adhesion molecules. The increased presence of TNF-α and IL-1β may synergistically amplify the expression of IL-6 and IL-8.
From the above, the question arises: Can exogenous pulmonary surfactant has a potential to act as adjuvant therapy in SARS-CoV-2 patients experiencing acute respiratory distress syndrome? [194]. Given the rapidly evolving nature and case severity of the current pandemic, we propose exogenous pulmonary surfactant therapy as worth an in depth investigation as an intervention in (early) cases of SARS-CoV-2 mediated acute respiratory distress syndrome. To the best of our knowledge, readily available bovine/porcine surfactant preparations are not currently being employed in hospitalized patients and could perhaps result in improved overall outcomes and shorter average duration of ICU stay in inpatients with COVID-19.
In the lungs, ACE2 is found on cell surfaces of airway epithelia and type 2 pneumocytes. These cells are responsible for the production and secretion of pulmonary surfactant. The virus uses these cells for fulminant replication, leading to their destruction and impaired surfactant production in the host. No therapeutic approach is presently directed at mitigating the resulting alveolar collapse, reduction in FRC and predictable respiratory failure. Exogenous pulmonary surfactant therapy, while widely used for the treatment of neonatal respiratory distress syndrome (nRDS), has not yet been implemented for treatment of SARS-CoV-2 associated respiratory failure to the best of our knowledge. We believe it may have, in theory, an advantage in adult cases of acute respiratory distress syndrome (ARDS).
Severe cases of SARS-CoV-2 typically involve the development of ARDS followed by septic shock or specific organ dysfunction (e.g., acute kidney injury, fulminant myocarditis), leading to death. Besides other medical treatment, support is generally limited to failing organs such as the lungs. This is achieved with oxygenation support (high-flow oxygen and non-invasive positive pressure ventilation) prior to tracheal intubation and mechanical ventilation. Thus, ARDS from rapid type-2 pneumocyte destruction appears to be an appropriate target for therapy which could mitigate alveolar compromise and prevent progression to later stage syndromes. With current figures indicating that approximately 20% of patients with proven SARS-CoV-2 infections are hospitalized, of whom 25% require critical care, it cannot be overstated that reducing ICU length of stay is critical in decreasing the health system burden. There is a unique opportunity to approach this problem using readily available bovine/porcine surfactant preparations, at the appropriate doses, for compassionate-use exemption in expedited clinical trials. To this end we propose the exploration of repeated early surfactant therapy-via endotracheal aerosolisation at a dose of 300 mg/kg in conjunction with standard procedures and therapies in intubated patients.
One of the problems when interpreting blood gas results in patients with ALI/ARDS, is the well described effects of the FiO2 on the pulmonary shunt [195-198]. This phenomenon is explained by the concept of absorption atelectasis where the alveoli with a low FRC and high content of oxygen, progressively collapse as the oxygen is absorbed by the relative excess capillary blood flow. If the presence of insufficient and insoluble nitrogen, which acts as an alveolar “strut” is replaced by the elevated alveolar oxygen, the unstable alveoli volume further decreases and hence the calculated shunt increases. Recently Karbing et al. suggested that if the PaO2/FiO2 ratio is used, the FiO2 level at which the PaO2/FiO2 ratio is measured, should be defined when quantifying the effects of therapeutic interventions or when specifying diagnostic criteria for ALI/ARDS because of this dependence of this questionable indirect index of pulmonary shunt on the inspired oxygen [199]. These authors, as Coetzee et al. [198] were of the opinion oxygenation difficulties is better described using more objective parameters such as directly calculated pulmonary shunt and ventilation/perfusion mismatch [199].
Meng et al. [130] found that surfactant administration did not significantly improve the PaO2/FiO2 ratio of ARDS patients. This publication was based on a meta-analysis of studies using various installation techniques. However, unless the before and after results were obtained on a similar FiO2, the results are meaningless.
Three decades ago, Seeger et al. [91] pointed out that persistent atelectasis of surfactant-deficient and fibrin-loaded alveoli may represent a key event to trigger fibroblast proliferation and fibrosis in late ARDS. Considering the forgoing, it is reasonable to speculate that combined treatment with surfactant and fibrinolytic agents could be considered early in the course of patients with ALI/ARDS. The theoretical basis is the speculation that, if the fibrin loaded alveoli can be cleared and surfactant instilled, it may result in improved gas exchange and compliance mechanics. This is an untested hypothesis.
As highlighted earlier, despite diversity in viral causes of pneumonia and ALI/ARDS, the repeated finding of similar lung pathology features related to viruses, dictate that DAD occurs early and frequently. Moreover, there is good evidence to show that, as a minimum, some of the alveolar pathology is related to disruption of alveolar-capillary integrity and Type II cell injury and surfactant dysfunction [111,200]. Furthermore, as part of DAD there is intra-alveolar clot formation secondary to intra-alveolar fibrin deposition owing to alveolar capillary injury and leakage. Intravascular microvascular thrombosis can also occur and extravascular fibrin deposition promotes lung dysfunction and the acute inflammatory response in addition to the microvascular and arteriole clot formation [201]. Recently, Magro et al. examined a role for complement activation and microvascular thrombosis in cases of persistent, severe COVID-19, since it is reasoned that the respiratory distress accompanying a subset of severe COVID-19, may be distinct from classic ARDS. Initially, there is relatively well-preserved lung mechanics despite the severity of hypoxemia, characterized by acceptable respiratory compliance and high shunt fraction, and increasing recognition of systemic features of a hypercoagulable state in this disease. Therefore, they reasoned, pathology and pathophysiology of COVID-19 might differ from that of typical ARDS [202]. Contrary to other publications on COVID-19 related lung involvement, these authors found a pattern of COVID-19 pneumonitis with a microvascular injury syndrome which was predominantly a small vessel inflammatory vasculitis of the alveolar septal capillaries mediated by activation of complement pathways and accompanied by a pro-coagulant state in the absence of DAD with hyaline membranes, inflammation, and type II pneumocyte hyperplasia, all hallmarks of classic ARDS. A recent position paper from Italian Society on Thrombosis and Haemostasis stated that although there is no confirmed evidence as yet from the laboratory, it is plausible that the plasma of COVID-19 patients is hypercoagulable, as suggested by preliminary laboratory information and many clinical observations [203]. It stated that it is also possible that pulmonary embolism is already present in more severely ill COVID-19 patients before hospitalisation, thus explaining the reported ineffectiveness of prophylactic doses of heparins during their hospital stay. The hypothesis of improving the clinical outcome of COVID-19 patients by simple and inexpensive antithrombotic drugs is very attractive, but several issues need to be addressed and clarified before adopting an aggressive anticoagulation approach. These include the appropriate timing of start of treatment, and the type and dosage of drug, while the impact of concomitant medications that are often taken by these subjects should also be taken into consideration. Moreover, it should be noted that approximately 50% of patients who have died of COVID-19 in Italy had three or more comorbidities such as atrial fibrillation or ischaemic heart disease and chronic kidney disease often requiring anticoagulant or antiplatelet treatment. The management of these is particularly challenging due to the potential interactions of concomitant therapies such as direct oral anticoagulants.
It has been known for several decades that extravascular fibrin deposition promotes lung dysfunction and the acute inflammatory response in diverse forms of ALI [204]. Within 3 days of disease onset, disordered coagulation (increased procoagulant activity) and fibrinolysis (decreased) promote extravascular fibrin deposition manifesting as alveolar fibrin deposition and intravascular thrombosis or disseminated intravascular coagulation. The alveolar deposition of fibrin is a result of inhibition of urokinase plasminogen activator (uPA) by plasminogen activators or inhibition of plasmin by antiplasmins [201]. Furthermore, it is this deposition that characterizes acute lung injury and repair. The success of anticoagulant or fibrinolytic strategies designed to reverse the abnormalities of local fibrin turnover in acute lung injury supports the inference that abnormalities of coagulation, fibrinolysis, and fibrin deposition have a critical role in the pathogenesis of acute lung injury. One small study examined a role for plasminogen activators in patients with ARDS (trauma, sepsis) who failed ventilation and PEEP. The hypothesis was that disseminated intravascular coagulation initiates ARDS by occluding the pulmonary microcirculation with microclots. The patients responded with significant improvement in oxygenation and no bleeding occurred and clotting parameters remained normal [205,206].
As Idell wrote in 2001 “To many in the pulmonary or critical care community, these reports may be surprising and the approach unfamiliar”. Anticoagulants exert anti-inflammatory as well as anticoagulant properties which may be utilized to protect against lung injury and ARDS. It is therefore reasoned that an early intervention with surfactant replacement with adjunctive fibrinolytic or thrombolytic anticoagulant agents may alter the clinical course and phenotypical expression of the ALI/ARDS disease pattern [201,207].
The alternative hypothesis is that there is no common pathological response of the lung to diverse insults, and that one size (early surfactant therapy with or without adjunctive agents) does not fit all. It is becoming clear that understanding both the likely pathophysiology and identifying the subpopulation at risk is crucial to establishing the basis for targeted and appropriate treatment. Since surfactant dysfunction occurs early during direct ARDS, surfactant is theoretically an attractive adjuvant treatment with non-invasive and invasive ventilation support [208-210]. During the earlier phase of viral-induced ARDS, surfactant treatment may prevent or ameliorate development of DAD and progression to P-SILI in the spontaneously breathing, but increasingly dyspnoeic patient. Surfactant replacement might prevent the progression from ALI (Type L) to the low volume, low compliance de-recruited lungs of the ARDS 2 / Type H. Adjunctive treatment with aerosolized surfactant could be administered while a patient is receiving any form of NIV support. Once the patient is intubated during the phase of ARDS 2 / Type H, the additional challenge is to avoid iatrogenic ventilator-induced lung injury by employing lung protective ventilation strategies. In the intubated patient surfactant could be administered as liquid boluses [211-214].
Given the fibrin and protein rich alveolar flooding which occurs, there is data which show convincing evidence that nebulized heparin and streptokinase improves oxygenation and compliance. The drugs are also taken up by the capillaries and one can speculate that its beneficial effects could be achieved at the locus (microvasculature) where the clotting pathology occurs. Again the emphasis on nebulization as a feasible route to address elements of the known pathophysiology associated with ALI/ARDS [49].
Despite relatively unchanged lung pathology related to direct ARDS throughout the past century (1918-2020), clinicians still have limited curative options on which to rely. We have reviewed some of the pathology of ALI/ARDS, inter alia the loss of surfactant and microvascular pathology associated with ARDS. In the neonate the use of surfactant clearly has demonstrated advantage but uncertain and variable results in adults have relegated the use of surfactant in adult ARDS to an uncertainty. We speculate that a prospective randomized trial using early and repeated surfactant in adults is required to address the possible advantage of this proposed treatment. In addition there is reason to suggest that surfactant may not only have a benefit from a mechanical point of view, it perhaps may have beneficial effects on the local inflammatory process in the ARDS lung.
The increased clotting tendency associated with the MOF/D (and ARDS), and in particular its effect on the lung vasculature, has pulmonary mechanical effects (such as increased dead space) and could limit oxygenation via the right ventricular overload and uncoupling of the right ventricle with the elevated pulmonary artery elastance. Hence, active management of this should include early effective and titrated inhibition of the accelerated coalgulation and thrombolysis via nebulization which is a relatively new concept worth further exploration.
COVID-19 has unique features, such as the dissociation between oxygenation, CT image and saturation seen in some patients. However, it also has features which are no different from the well-defined ALI/ARDS pathology. Hence, the suggestion of surfactant use (amongst other therapies) and effective anticoalgulation / thrombolysis equally applies to COVID-19.
Clinicians often are confronted with confusing facts and even disinformation. The best way to navigate a safe course is to rely on the known pathology and devise methods to specifically circumnavigate those with targeted therapy. The latter often, in ARDS, is not curative but may support and buy time for the patient to recover from the complex pathology.
No specific curative pharmacotherapies have been identified in patients with ALI/ARDS. The caveat is that these patients should be identified early at onset of disease [85].
Not Applicable
Johann M van Zyl and Johan Smith are designers and patent holders of the synthetic lung surfactant Synsurfâ mentioned in this review. This surfactant was patented by InnovUS of Stellenbosch University. The authors declare no competing interest.
- 1. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, et al. (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367: 1260-1263. [Crossref]
- 2. Geng YJ, Wei ZY, Qianb HY, Huang J, Lodatoa R, et al. (2020) Pathophysiological characteristics and therapeutic approaches for pulmonary injury and cardiovascular complications of coronavirus disease. Cardiovasc Pathol 47: 107228. [Crossref]
- 3. Xu H, Zhong L, Deng J, Peng J, Dan H, et al. (2020) High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 12: 8.
- 4. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, et al. (2020) COVID-19 does not lead to a “typical” acute respiratory distress syndrome. Am J Resp Crit Care Med 201:1299-1300. [Crossref]
- 5. Gattinoni L, Chiumello D, Rossi S (2020) COVID-19 Pneumonia: ARDS or not? Crit Care 24: 154. [Crossref]
- 6. Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, et al. (2020) Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc 22: 95-97. [Crossref]
- 7. Nieman GF, Al-Khalisy H, Kollisch-Singule M, Satalin J, Blair S, et al. (2020) A physiologically informed strategy to effectively open, stabilize, and protect the acutely injured lung. Front Physiol 11: 227. [Crossref]
- 8. Murray J, Matthay M, Luce J, Flick M (1988) An expanded definition of the adult respiratory syndrome. Am Rev Resp Dis 138: 720-723. [Crossref]
- 9. Ranieri M, Rubenfeld G, Thompson T, Ferguson N, Cladwell E, et al. (2012) Acute respiratory distress syndrome: The Berlin definition. JAMA 307: 2526-2533. [Crossref]
- 10. Robbins, Cotran (1999) Pathological basis of disease. Ed. V Kumar, A Abbas, N Fausto. Elsevier Saunders, Philadephia, p751.
- 11. Karkhanis VS, Joshi JM (2012) Pleural effusion: diagnosis, treatment and management. Open Access Emerg Med 4: 31-52. [Crossref]
- 12. Tang NL, Chan PK, Wong CK, To KF, Wu AK, et al. (2005) Early enhanced expression of interferon-inducible protein–10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin Chem 51: 2333-2340. [Crossref]
- 13. Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, et al. (2003) Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol 34: 743-748. [Crossref]
- 14. Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, et al. (2005) Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 18: 1-10. [Crossref]
- 15. Wolbach SB, Frothingham C (1923) The influenza epidemic at camp Devens in 1918. A study of the pathology of the fatal cases. Arch Int Med (Chic) 32: 571- 600.
- 16. Taubenberger JK, Morens DM (2008) The pathology of influenza virus infections. Annu Rev Pathol 3: 499-522. [Crossref]
- 17. Bellingan GJ (2002) The pulmonary physician in critical care 6: The pathogenesis of ALI/ARDS. Thorax 57: 540-546. [Crossref]
- 18. Katzenstein ALA, Bloor CM, Liebow AA (1996) Diffuse alveolar damage-the role of oxygen, shock and related factors. Am J Pathol 85: 209-228. [Crossref]
- 19. LeCount ER (1919) Disseminated necrosis of the pulmonary capillaries in influenza pneumonia. JAMA 72: 1519-1520.
- 20. Thille AW, Esteban A, Fernández-Segoviano P, Rodriguez JM, Aramburu JA, et al. (2013) Chronology of histological lesions in acute respiratory distress syndrome with diffuse alveolar damage: a prospective cohort study of clinical autopsies. Lancet Respir Med 1: 395-401. [Crossref]
- 21. Katzenstein AL, Bloor CM, Leibow W (1976) Diffuse alveolar damage -the role of oxygen, shock and related factors. A Review. Am J Pathol 85: 2019-228.
- 22. Beasly MB (2010) The pathologists approach to lung injury. Arch Pathol Lab Med 134: 719-727. [Crossref]
- 23. Tomashefski JF (2000) Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med 21: 435-466. [Crossref]
- 24. Kuiken T, Taubenberger J (2008) Pathology of human influenza revisited. Vaccine 1226: D59-D66. [Crossref]
- 25. Louria DB, Blumenfeld HL, Ellis JT, Kilbourne ED, Rogers DE (1959) Studies on Influenza in the pandemic of 1957-1958. II. Pulmonary complications of Influenza. J Clin Invest 38: 213-265. [Crossref]
- 26. To KF, Chan PK, Chan KF, Lee WK, Lam WY, et al. (2001) Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol 63: 242-246. [Crossref]
- 27. Wang W, Dong C, Hu Y, Li, C, Ren Q (2020) Temporal changes of CT findings in 90 patients with COVID-19 pneumonia: A longitudinal study. Radiology 296: E55-E64. [Crossref]
- 28. Nin N, Sánchez-Rodrígueza C, Ver LS, Cardinal P, Ferruelo A, et al. (2012) Lung histopathological findings in fatal pandemic influenza A (H1N1). Med Intensiva 36: 24-31. [Crossref]
- 29. Alsaad KO, Hajeer AH, Al Balwi M, Al Moaiqel M, Al Oudah N, et al. (2018) Histopathology of middle east respiratory syndrome coronovirus (MERS-CoV) infection - clinicopathological and ultrastructural study. Histopathology 72: 516-524. [Crossref]
- 30. Ng DL, Al Hosani F, Keating MK, Gerber SI, Jones TL, et al. (2016) Clinicopathologic, immunohistochemical, and ultrastructural findings of a fatal case of middle east respiratory syndrome coronavirus infection in the United Arab Emirates. Am J Pathol 186: 652-658. [Crossref]
- 31. Xu Z, Shi L, Wang Y, Zhang J, Huang L, et al (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Resp Med 8: 420-422. [Crossref]
- 32. Tian S, Xiong Y, Liu H, Niu L, Guo J, et al. (2020) Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol 33: 1007-1014.
- 33. Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, et al. (2020) Post‐mortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology 77: 198-209. [Crossref]
- 34. Zhang T, Sun LX, Feng RE (2020) Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019. Zhonghua Jie He He Hu Xi Za Zhi 43: 496-502. [Crossref]
- 35. Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, et al. (2020) Pulmonary post-mortem findings in a large series of COVID-19 cases from Northern Italy. medRxiv: 20054262.
- 36. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, et al. (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 383: 120-128. [Crossref]
- 37. Cardinal-Fernández P, Lorente JA, Ballén-Barragán A, Matute-Bello G (2017) Acute respiratory distress syndrome and diffuse alveolar damage. New insights on a complex relationship. Ann Am Thorac Soc 14: 844-850. [Crossref]
- 38. Sweeney RM, McAuley DF (2016) Acute respiratory distress syndrome. Lancet 388: 2416-2430.
- 39. Gurka D, Balk R (2007) Acute Respiratroy Failure In: Critical Care Medicine: Principles of diagnosis and management in the adult patient. 3rd Ed. eds., J Parillo, P Dellinger. Mosby Elsevier, Philadephia 1008: 773-794.
- 40. Artigas A, Le Gall JR, Carlet J, Blnach L, Fernadez R. Clinical presentation, prognostic factors and outcome of ARDS in the European Collaberative Study (1985-1987). In: Adult Respiratory Distress Syndrome. Zapol, W. and Lemaire, F. eds., 1991. Lung Biology in Health and Disease, Marcel Dekker, New York. pp.37- 63.
- 41. Repine JE. Evidence implicating neutrophils in the development of ARDS. In Acute Respiratory Failure. Zapol, W. and Falke, K. eds., 1985. Lung Biology in Health and Disease, Marcel Dekker, New York pp.347-377.
- 42. Petty T (1994) ARDS: Historical perspective. Chest 105: 445-457.
- 43. Fry D (1992) In: Multiple System Organ Failure. Mosby, St Louis pp.3-14.
- 44. Hudson LD, Milberg JA, Anardi D, Maunder RJ (1995) Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med 151: 293-301. [Crossref]
- 45. Jones R, Zapol W, Tomashefski J, Kirton O, Kobayahi K. Pulmonary vascular pathology: Human and experimental studies. In: Acute Respiratory Failure. Zapol, W. and Falke, K. eds., 1985 Lung Biology in Health and Disease. Marcel Dekker, New York pp.23-160.
- 46. Villar J, Kacmarek RM, Hedenstierna G (2004) From ventilator-induced lung injury to physician-induced lung injury: why the reluctance to use small tidal volumes? Acta Anaesth Scand 48: 267-271. [Crossref]
- 47. Lemaire F, Harf A, Teisseirie BP. Oxygen exchange across the acutely injured lung. In: Acute Respiratory Failure. Zapol, W. and Falke, K. eds., 1985. Lung Biology in Health and Disease, Marcel Dekker, New York pp.5121-553.
- 48. Jolin A, Bjertnaes A (1991) Hypoxic pulmonary vasoconstriction in ARDS. Acta Anaesth Scand 35:40-54.
- 49. Raurich JM, Vilar M, Colomar A, Ibanez J, Ayestaran I, et al. (2010) Prognostic value of the pulmonary dead-space fraction during early and intermediate phases of acute respiratory distress syndrome. Respir Care 55: 282-287. [Crossref]
- 50. Ospina-Tascón GA, Bautista DF, Madrińán HJ, Quińones WF, Calderón-Tapia LE, et al. (2020) Microcirculatory dysfunction and dead-space ventilation in early ARDS: a hypothesis-generating observational study. Ann Intensive Care 10: 35. [Crossref]
- 51. Fourie P (1989) The evaluation of cardiac function with reference to pulmonary hypertension, Stellenbosch University.
- 52. Mahmoud AAAM, Mahmoud HE, Mahran MA, Khaled M (2020) Streptokinase versus unfractionated heparin nebulization in patients with severe acute respiratory distress syndrome (ARDS): A randomized controlled trial with observational controls. J Cardiothorac Vasc Anesth 34: 436-443. [Crossref]
- 53. Beydon L, Lemaire F, Jonson B. Lung mechanics in ARDS: Compliance and pressure-volume curves. In: Adult Respiratroy Distress Syndrome. Zapol, W. and Lemaire, F. eds., 1991. Lung Biology in Health and Disease, Marcel Dekker, New York. 50: 139-156.
- 54. Fan E, Brodie D, Slutsky AS (2018) Acute Respiratory Distress Syndrome Advances in Diagnosis and Treatment. JAMA 319: 698-710. [Crossref]
- 55. Brower RG, Matthay MA, Morris A, Schoenfeld D, Tompson BT (2000) The acute respiratory distress syndrome network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N EngI J Med 342: 1301-1308. [Crossref]
- 56. Cheung OY, Chan JWM, Ng CK, Koo CK (2004) The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology 45: 119-124. [Crossref]
- 57. Sweeney RM, McAuley DF (2016) Acute respiratory distress syndrome. Lancet 388: 2416-2430. [Crossref]
- 58. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E (2012) Acute respiratory distress syndrome: The Berlin definition. JAMA 307: 2526-2533. [Crossref]
- 59. Petty TL, Ashbaugh DG (1971) The adult respiratory distress syndrome. Chest 60: 233-239. [Crossref]
- 60. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE (1967) Acute respiratory distress in adults. Lancet 2: 319-323. [Crossref]
- 61. Coetzee A, Swanevelder C, Van der Spuy G, Jansen J (1995) Gas exchange indices- how valid are they? S Afr Med J 85: 1227-1232. [Crossref]
- 62. Meduri GU, Annane D, Chrousos GP, Marik PE, Sinclair SE (2009) Activation and regulation of systemic inflammation in ARDS rationale for prolonged glucocorticoid therapy. Chest 136: 1631-1643. [Crossref]
- 63. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A (2020) Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27: 992-1000. [Crossref]
- 64. Papazian L, Aubron L, Cliché J, Combes A, Dreyfuss D, et al. (2019) Formal guidelines: Management of acute respiratory distress syndrome. Ann Int Care 9: 69. [Crossref]
- 65. Bellami G, Laffey J, Pham T, Fan E, Brochard L, et al. (2016) Epidemiology patterns of care and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315: 788-800. [Crossref]
- 66. Amato M, Meade M, Slutsky A, Brochard L, Costa ELV, et al. (2015) Driving pressure and survival in the acute respiratory distress syndrome. N EngI J Med 37: 747-755. [Crossref]
- 67. Hickling K, Walsh J, Henderson S, Jackson R (1994) Low mortality rate in acute respiratory distress syndrome using low volume pressure limited ventilation. A prospective study. Crit Care Med 22: 1568-1578. [Crossref]
- 68. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN (2020) The pathophysiology of “happy” hypoxemia in COVID-19. Respir Res 21: 198. [Crossref]
- 69. Huang C, Wang Y, Li X, Ren L, Zhao J, et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497-506.
- 70. Merad M, Martin JC (2020) Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20: 1-8. [Crossref]
- 71. Light RB (1999) Pulmonary pathology in pneumococcal pneumonia. Sem Resp Infection 14: 218-226.
- 72. Dunham-Snary K, Wu D, Sykes E, Thakrar A, Parlow LRG, et al. (2017) Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 151:181-192. [Crossref]
- 73. Lumb A, Slinger P (2015) Hypoxic pulmonary vasoconstriction. Physiology and anesthetic implications. Anesthesiology 122: 932-946. [Crossref]
- 74. Luyt S, Coetzee A, Lahner D, Jansen J (1997) Nitrous oxide has little effect on right ventricle function and pulmonary artery pressure in patients during acute respiratory distress syndrome. S Afr Med J 87: 639-642. [Crossref]
- 75. Brochard L, Slutsky A, Pesenti A (2017) Mechanical ventilation to minimize progression of lung injury in acute respiratory failure. Am J Respir Crit Care Med 195: 438-442. [Crossref]
- 76. Zapol W (1992) Volotrauma and the oxygenator in patients with ARDS. Anesthesiology 77: 847-849. [Crossref]
- 77. Festic E, Bansal V, Kor DJ, Gajic O (2015) SpO2/FiO2 ratio on hospital admission is an indicator of early acute respiratory distress syndrome development among patients at risk. J Intensive Care Med 30: 209-216. [Crossref]
- 78. Villar J, Kacmarek RM, Hedenstierna G, (2004) From ventilator-induced lung injury to physician-induced lung injury: why the reluctance to use small tidal volumes? Acta Anaesth Scand 48: 267-271. [Crossref]
- 79. Gattinoni L, Caironi P, Cressoni M, Chiumello D, Ranieri VM, et al. (2006) Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med 354: 1775-1786. [Crossref]
- 80. Antonelli M, Conti G, Rocco M, Bufi M, De Blasi RA, et al. (1998) A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 339: 429-435. [Crossref]
- 81. Frat JP, Thille AW, Mercat A, Girault C, Ragot S, et al. (2015) High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med 372: 2185-2196.
- 82. Brochard L, Lefebvre JC, Cordioli RL, Akoumianaki E, Richard JCM, et al. (2014) Noninvasive ventilation for patients with hypoxemic acute respiratory failure. Semin Respir Crit Care Med 35: 492-500. [Crossref]
- 83. Jaber S, Lescot T, Futier E, Paugam-Burtz C, Seguin P, et al. (2016) Effect of noninvasive ventilation on tracheal reintubation among patients with hypoxemic respiratory failure following abdominal surgery: A randomized clinical trial. JAMA 315: 1345-1353. [Crossref]
- 84. Patel BK, Wolfe KS, Pohlman AS, Hall JB, Kress JP, (2016) Effect of noninvasive ventilation delivered by helmet vs face mask on the rate of endotracheal intubation in patients with acute respiratory distress syndrome: A randomized clinical trial. JAMA 315: 2435-2441. [Crossref]
- 85. Raghavendran K, Willson D, Notter RH, (2011) Surfactant therapy of ALI and ARDS. Crit Care Clin 27: 525-559. [Crossref]
- 86. Hand IL, Shepard EK, Krauss AN, Auld PAM, (1990) Ventilation-perfusion abnormalities in the preterm infant with hyaline membrane disease: A two-compartment model of the neonatal lung. Pediatr Pulmonol 9: 206-213. [Crossref]
- 87. Spragg R, Gilliard N, Richman P, Smith RM, Hite RD, et al. (1994) Acute effects of a single dose of porcine surfactant on patients with the adult respiratory distress syndrome. Chest 105: 195-202. [Crossref]
- 88. Pang C, Su M, Jiang T (2017) Efficacy of pulmonary surfactant in the treatment of adult respiratory distress syndrome (ARDS). Biomed Res 28: 9801-9804.
- 89. Kribs A, Vierzig A, Hünseler C, Eifinger F, Welzing L, (2008) Early surfactant in spontaneously breathing with nCPAP in ELBW infants - a single centre four-year experience. Acta Paediatrica 97: 293-298. [Crossref]
- 90. Ghadiali SN, Gaver DP, (2008) Biomechanics of liquid-epithelium interactions in pulmonary airways. Respir Physiol Neurobiol 163: 232-243.[Crossref]
- 91. Seeger W, Gunther A, Walmrath, HD, Grimminger F, Lasch HG (1993) Alveolar surfactant and adult respiratory distress syndrome. Pathogenic role and therapeutic prospects. Clin Investig 71:177-190. [Crossref]
- 92. Rojas-Reyes MX, Morley CJ, Soll R (2012) Prophylactic versus selective use of surfactant in preventing morbidity and mortality in preterm infants. Cochrane Database Syst Rev 3: CD000510. [Crossref]
- 93. Bahadue FL, Soll R (2012) Early versus delayed selective surfactant treatment for neonatal respiratory distress syndrome. Cochrane Database Syst Rev 11: CD001456. [Crossref]
- 94. Auten RL, Notter RH, Kendig JW, Davis JM, Shapiro DL (1996) Surfactant treatment of full-term newborns with respiratory failure. Pediatrics 87: 101-107. [Crossref]
- 95. Findlay RD, Taeusch HW, Walther FJ (1996) Surfactant replacement therapy for meconium aspiration syndrome. Pediatrics 97: 48-52. [Crossref]
- 96. Lotze A, Mitchell BR, Bulas DI, Zola EM, Shalwitz RA, et al. (1998) Multicenter study of surfactant (beractant) use in the treatment of term infants with severe respiratory failure. J Pediatr 132: 40-47. [Crossref]
- 97. Khammash H, Perlman M, Wojtulewicz J, Dunn M (1993) Surfactant therapy in full-term neonates with severe respiratory failure. Pediatrics 92: 135-139. [Crossref]
- 98. Jat KR, Chawla D (2015) Surfactant therapy for bronchiolitis in critically ill infants. Cochrane Database Syst Rev 8: CD009194. [Crossref]
- 99. Luchetti M, Ferrero F, Gallini C, Natale A, Pigna A, et al. (2002) Multicenter, randomized, controlled study of porcine surfactant in severe respiratory syncytial virus-induced respiratory failure. Pediatr Crit Care Med 3:261-268.[Crossref]
- 100. Bates JHT, Smith BJ, Allen GB (2015) Computational Models of Ventilator Induced Lung Injury and Surfactant Dysfunction. Drug Discov Today Dis Models 15:17-22. [Crossref]
- 101. Sharma V, Berkelhamer S, Lakshminrusimha S (2015) Persistent pulmonary hypertension of the newborn. Matern Health Neonatal Perinat: s40748-015-0015-4
- 102. Rebello M, Jobe AH, Eisele IW, Ikegami M (1996) Alveolar and tissue surfactant pool sizes in humans. Am J Respir Crit Care Med 154: 625-628. [Crossref]
- 103. Fessler MB, Summer RS, (2016) Surfactant lipids at the host–environment interface metabolic sensors, suppressors, and effectors of inflammatory lung disease. Am J Respir Cell Mol Biol 54: 624-635. [Crossref]
- 104. Kuronuma K, Mitsuzawa H, Takeda K, Nishitani C, Chan ED (2009) Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J Biol Chem 284: 25488-25500. [Crossref]
- 105. Numata M, Chu HW, Dakhama A, Voelker DR (2010) Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc Natl Acad Sci 107: 320-325. [Crossref]
- 106. Voelker DR, Numata M (2019) Phospholipid regulation of innate immunity and respiratory viral infection. J Biol Chem 294: 4282-4289. [Crossref]
- 107. Brown ES, Johnson RP, Clements JA (1959) Pulmonary surface tension. J Appl Physiol 14: 17-20. [Crossref]
- 108. Watkins J (1968) The surface properties of pure phospholipids in relation to those of lung extracts. Biochim Biophys Acta 152: 293-306. [Crossref]
- 109. Pré J, Perret G, Bladier D (1983) Lecithin content estimate of human alveolar lining layer: Comparison with mouse, rat and rabbit. Comparative Biochemistry and Physiology Part A: Physiology 76: 393-395.
- 110. Lewis JF, Jobe AH (1993) Surfactant and the adult respiratory distress syndrome. Am Rev Respir Dis 147: 218-233.[Crossref]
- 111. Hallman M, Spragg R, Harrell JH, Moser KM, Gluck L (1982) Evidence of lung surfactant abnormality in respiratory failure. Study of bronchoalveolar lavage phospholipids, surface activity, phospholipase activity, and plasma myoinositol. J Clin Invest 70: 673-683. [Crossref]
- 112. Petty TL, Reiss OK, Paul GW, Silvers GW, Elkins ND (1977) Characteristics of pulmonary surfactant in adult respiratory syndrome associated with trauma and shock. Am Rev Respir Dis 115: 531-536. [Crossref]
- 113. Petty TL, Silvers GW, Paul GW, Stanford RE (1979) Abnormalities in lung elastic properties and surfactant function in adult respiratory distress syndrome. Chest 75: 571-574. [Crossref]
- 114. Gregory TW, Longmore M, Moxley JA Whitsett CR, Reed AA, et al. (1991) Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J Clin Invest 65: 1976-1981. [Crossref]
- 115. Echaide M, Autilio C, Arroyo R, Perez-Gil J (2017) Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochim Biophys Acta Biomembr 1859:1725-1739. [Crossref]
- 116. Seeger W, Pison U, Buchhorn R, Obertacke U Joka T (1990) Treatment of respiratory distress syndrome. Surfactant abnormalities and adult respiratory failure. Lung 168: 891-902.
- 117. Günther A, Ruppert C, Schmidt R, Markart P, Grimminger F, et al. (2001) Surfactant alteration and replacement in acute respiratory distress syndrome. Respir Res 2: 353-364. [Crossref]
- 118. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, et al. (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395:1033-1034. [Crossref]
- 119. Dushianthan A, Goss V, Cusack R, Grocott MPW, Postle AD (2014) Phospholipid composition and kinetics in different endobronchial fractions from healthy volunteers. BMC Pulm Med14:10. [Crossref]
- 120. Sebag SC, Bastarache JA, Ware LB (2011) Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr Pharm Biotechnol 12: 1481-1496. [Crossref]
- 121. Schmidt R, Markart P, Ruppert C, Wygrecka M, Kuchenbuch T, et al. (2007) Time-dependent changes in pulmonary surfactant function and composition in acute respiratory distress syndrome due to pneumonia or aspiration. Respir Res 8: 55. [Crossref]
- 122. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, et al. (2020) COVID‑19 pneumonia: different respiratory treatments for different phenotypes? Intensive Care Med 46: 1099-1102. [Crossref]
- 123. Touqui L, Arbibe L (1999) A role for phospholipase A2 in ARDS pathogenesis. Mol Med Today 5: 244- 249. [Crossref]
- 124. Niewoehner DE, Rice K, Duane P, Sinha AA, Gebhard R, et al. (1989) Induction of alveolar epithelial injury by phospholipase A2. J Appl Physiol 66: 261-267. [Crossref]
- 125. Kakuta Y, Sasaki H, Takishima T (1991) Effect of artificial surfactant on ciliary beat frequency in guinea pig trachea. Respir Physiol 83: 313-321. [Crossref]
- 126. Willson DF, Notter RH (2011) The future of exogenous surfactant therapy. Respir Care 56:1369-1386. [Crossref]
- 127. Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342:1334-1349. [Crossref]
- 128. Lewis JF, Veldhuizen RAW (2006) The Future of Surfactant Therapy during ALI/ARDS. Semin Respir Crit Care Me 4: 377-388. [Crossref]
- 129. Davidson WJ, Dorscheid D, Spragg R, Schulzer M, Mak E, et al. (2006) Exogenous pulmonary surfactant for the treatment of adult patients with acute respiratory distress syndrome: results of a meta-analysis. Crit Care 10: R41. [Crossref]
- 130. Meng SS, Chang W, Lu ZH, Xie F, Qiu HB, et al. (2019) Effect of surfactant administration on outcomes of adult patients in acute respiratory distress syndrome: a meta-analysis of randomized controlled trials. BMC Pulm Med 19: 9. [Crossref]
- 131. Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, et al. (1996) Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. N Engl J Med 334: 1417-1422. [Crossref]
- 132. Spragg RG, Taut FJH, Lewis JF, Schenk P, Ruppert C, et al. (2011) Recombinant surfactant protein C-based surfactant for patients with severe direct lung injury. Am J Resp Crit Care Med 183: 1055-1061. [Crossref]
- 133. Willson DF, Truwit JD, Conaway MR, Traul CS, Egan EE (2015) The adult calfactant in acute respiratory distress syndrome trial. Chest 148: 356-364. [Crossref]
- 134. Taut FJ, Rippin G, Schenk P, Findlay G, Wurst W, et al. (2008) A Search for subgroups of patients with ARDS who may benefit from surfactant replacement therapy: a pooled analysis of five studies with recombinant surfactant protein C surfactant (Venticute). Chest 134:724-732. [Crossref]
- 135. Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, et al. (2004) Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med 351: 884-892. [Crossref]
- 136. Kesecioglu J, Beale R, Stewart TE, Findlay GP, Rouby JJ, et al. (2009) Exogenous natural surfactant for treatment of acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 180: 989-994. [Crossref]
- 137. Dizdar EA, Sari FN, Aydemir C, Oguz SS, Erdeve O, et al. (2012) A randomized, controlled trial of poractant alfa versus beractant in the treatment of preterm infants with respiratory distress syndrome. Am J Perinatol 29: 95-100. [Crossref]
- 138. Ramanathan R, Bhatia JJ, Sekar K, Ernst FR (2013) Mortality in preterm infants with respiratory distress syndrome treated with poractant alfa, calfactant or beractant: a retrospective study. J Perinatol 33: 119-125. [Crossref]
- 139. Sinha SK, Lacaze-Masmonteil T, Soler AV, Wiswell TE, Gadzinowski J, et al. (2005) A multicenter,randomized, controlled trial of lucinactant versus poractant alfa among very premature infants at high risk for respiratory distress syndrome. Pediatrics 115: 1030-1038. [Crossref]
- 140. Dunn MS, Shennan AT, Zayack D, Possmayer F (1991) Bovine surfactant replacement therapy in neonates of less than 30 weeks' gestation: A randomized controlled trial of prophylaxis versus treatment. Pediatrics 87:377-386. [Crossref]
- 141. Sweet DG, Turner MA, Straňák Z, Plavka R, Clarke P, et al. (2017) A first-in-human clinical study of a new SP-B and SP-C enriched synthetic surfactant (CHF5633) in preterm babies with respiratory distress syndrome. Arch Dis Child Fetal Neonatal Ed 102: F497-F503. [Crossref]
- 142. El Shahed AI, Dargaville PA, Ohlsson A, Soll R (2014) Surfactant for meconium aspiration syndrome in term and late preterm infants. Cochrane Database Syst Rev 12: CD002054. [Crossref]
- 143. Moya FR, Gadzinowski J, Bancalari E, Salinas V, Kopelman B, et al. (2005) A multicenter, randomized, masked, comparison trial of lucinactant, colfosceril palmitate, and beractant for the prevention of respiratory distress syndrome among very preterm infants. Pediatrics 115:1018-1029. [Crossref]
- 144. Spragg RG, Lewis JF, Wurst W, Häfner D, Bäughman RP, et al. (2003) Treatment of acute respiratory distress syndrome with recombinant surfactant protein C surfactant. Am J Respir Crit Care Med 167:1562- 1566. [Crossref]
- 145. Walther FJ (2008) Synthetic Lung Surfactant Optimized for Biomedical Application.
- 146. Seehase M, Collins JJP, Kuypers E, Jellema RK, Ophelders DRMG, et al. (2012) New surfactant with SP-B and C analogs gives survival benefit after inactivation in preterm lambs. PLoS One 7: e47631. [Crossref]
- 147. Van Rensburg L, Van Zyl JM, Smith J, Goussard P (2019) Effect of exogenous surfactant on paediatric bronchoalveolar lavage derived macrophages’ cytokine secretion. BMC Pulm Med 19: 236.
- 148. Van Zyl JM, Smith J (2013) Surfactant treatment before first breath for respiratory distress syndrome in preterm lambs: comparison of a peptide-containing synthetic lung surfactant with porcine-derived surfactant. Drug Des Devel Ther 7: 905-916. [Crossref]
- 149. Van Rensburg L, Van Zyl JM, Smith J (2020) Comparative study of pulmonary surfactants: Immunoactive properties of Synsurf, Curosurf and Liposurf. Cur Top Pharmacol 24: 27-47.
- 150. Ahlström JZ, Massaro F, Mikolka P, Feinstein R, Perchiazzi G, et al. (2019) Synthetic surfactant with a recombinant surfactant protein C analogue improves lung function and attenuates inflammation in a model of acute respiratory distress syndrome in adult rabbits. Respir Res 20: 245. [Crossref]
- 151. Lin WH, Cramer SG, Turcotte JG, Thrall R S (1997) A diether phosphonolipid surfactant analog, DEPN-8, is resistant to phospholipase-C cleavage. Respiration 64: 96-101.[Crossref]
- 152. Bautin A, Khubulava G, Kozlov I, Poptzov V, Osovskikh V, et al. (2006) Surfactant therapy for patients with ARDS after cardiac surgery. J Liposome Res16: 265-272. [Crossref]
- 153. Gregory TJ, Steinberg KP, Spragg R, Gadek JE, Hyers TM, et al. (1997) Bovine surfactant therapy for patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 155: 1309-1315. [Crossref]
- 154. Grotberg JB, Filoche M, Willson DF, Raghavendran K, Notter RH (2017) Did reduced alveolar delivery of surfactant contribute to negative results in adults with acute respiratory distress syndrome? Am J Resp Crit Care Med 195: 538-540. [Crossref]
- 155. Filoche M, Tai CF, Grotberg JB (2015) Three-dimensional model of surfactant replacement therapy. Proc Natl Acad Sci USA 112: 9287-9292. [Crossref]
- 156. Gunther A, Schmidt R, Harodt J, Schmehl T, Walmrath D, et al. (2002) Bronchoscopic administration of bovine natural surfactant in ARDS and septic shock: impact on biophysical and biochemical surfactant properties. Eur Respir J 19: 797-804. [Crossref]
- 157. Walmrath D, Grimminger F, Pappert D, Knothe C, Obertacke U, et al. (2002) Bronchoscopic administration of bovine natural surfactant in ARDS and septic shock: impact on gas exchange and haemodynamics. Eur Respir J 19: 805-810. [Crossref]
- 158. Hallman M, Merritt TA, Pohjavuori M, Gluck L (1986) Effect of surfactant substitution on lung effluent phospholipids in respiratory distress syndrome: evaluation of surfactant phospholipid turnover, pool size, and the relationship to severity of respiratory failure. Pediatr Res 20: 1228-1235. [Crossref]
- 159. Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, et al. (1999) Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med 160: 1843-1850. [Crossref]
- 160. Jacobson W, Park GR, Saich T, Holcroft J (1993) Surfactant and adult respiratory distress syndrome. Br J Anaesth 70: 522-526.
- 161. Pettenazzo A, Ikegami M, Seidner S, Jobe A (1988) Clearance of surfactant phosphatidylcholine from adult rabbit lungs. J Appl Physiol 64: 120-127. [Crossref]
- 162. Mols G, Priebe HJ, Guttmann J (2006) Alveolar recruitment in acute lung injury. Br J Anaesth 96: 156-166. [Crossref]
- 163. Krause MF, Jäkel C, Haberstroh J, Schulte-Mönting J, Leititis JU, et al. (2001) Alveolar recruitment promotes homogeneous surfactant distribution in a piglet model of lung injury. Pediatr Res 50: 34-43. [Crossref]
- 164. Tsangaris I, Galiatsou E, Kostanti E, Nakos G (2007) The effect of exogenous surfactant in patients with lung contusions and acute lung injury. Int Care Med 33: 851-855. [Crossref]
- 165. Nakamura CT, Ripka JF, McVeigh K, Kapoor N, Keens TG (2001) Bronchoscopic instillation of surfactant in acute respiratory distress syndrome. Pediatr Pulmonol 31: 317-332. [Crossref]
- 166. Friedrich I, Börgermann J, Splittgerber FH, Brinkmann M, Reidemeister JC, et al. (2004) Bronchoscopic surfactant administration preserves gas exchange and pulmonary compliance after single lung transplantation in dogs. J Thorac Cardiovasc Surg 127: 335-343.[Crossref]
- 167. Darquenne C (2012) Aerosol Deposition in Health and Disease. J Aerosol Med Pulm Del 25: 140-147. [Crossref]
- 168. Laube BL, Janssens HM, de Jongh FHC, Devadason SG, Dhand R, et al. (2011) What the pulmonary specialist should know about the new inhalation therapies. Eur Respir J 37: 1308-1331. [Crossref]
- 169. Pritchard JN, Hatley RHM, Denyer J, von Hollen D (2018) Mesh nebulizers have become the first choice for new nebulized pharmaceutical drug developments. Ther Deliv 9:121-136. [Crossref]
- 170. Bianco F, Ricci F, Catozzi C, Murgai X, Schlun M, et al. (2019) From bench to bedside: in vitro and in vivo evaluation of a neonate-focused nebulized surfactant delivery strategy. Resp Res 20: 134. [Crossref]
- 171. Porra L, Dégrugilliers L, Broche L, Albu G, Strengell S, et al. (2018) Quantitative imaging of regional aerosol deposition, lung ventilation and morphology by synchrotron radiation CT. Scientific Reports 8: 3519.
- 172. Sinclair SE, Polissar NL, Altemeier WA (2010) Spatial distribution of sequential ventilation during mechanical ventilation of the uninjured lung: an argument for cyclical airway collapse and expansion. BMC Pulm Med 10: 25. [Crossref]
- 173. Kim HC, Won YY (2018) Clinical, technological, and economic issues associated with developing new lung surfactant therapeutics. Biotechnol Adv 4: 1185-1193. [Crossref]
- 174. Lewis JF, McCaig L (1993) Aerosolized versus instilled exogenous surfactant in a nonuniform pattern of lung injury. Am Rev Respir Dis 148: 1187-1193. [Crossref]
- 175. Walther FJ, Hernandez-Juviel JM, Waring AJ (2014) Aerosol delivery of synthetic lung surfactant. Peer J 2: e403. [Crossref]
- 176. Agenbag A (2017) Biophysical properties of experimental compositions of a synthetic pulmonary surfactant synsurf for aerosolation. Thesis (MSc) Stellenbosch University.
- 177. Bachofen H, Schürch S (2001) Alveolar surface forces and lung architecture. Comp Biochem Physiol A Mol Integr Physiol 129: 183-193. [Crossref]
- 178. Ghadiali SN, Gaver III DP (2003) The influence of non-equilibrium surfactant dynamics on the flow of a semi-infinite bubble in a rigid cylindrical capillary tube. J Fluid Mech 478: 165-196. [Crossref]
- 179. Naire S, Jensen OE (2005) Epithelial cell deformation during surfactant-mediated airway reopening: a theoretical model. J Appl Physiol 99: 458-471. [Crossref]
- 180. Halpern D, Gaver III DP (2012) The influence of surfactant on the propagation of a semi-infinite bubble through a liquid-filled compliant channel. J Fluid Mech 698: 125-159. [Crossref]
- 181. Stebe KJ, Barthes-Biese lD (1995) Marangoni effects of adsorption–desorption-controlled surfactants on the leading edge of an infinitely long bubble in a capillary. J Fluid Mech 286: 25-48.
- 182. Smith BJ, Lukens S, Yamaguchi E, Gaver III DP (2011) Lagrangian transport properties of pulmonary interfacial flows. J Fluid Mech 705: 234-257. [Crossref]
- 183. Zimmer ME, Williams HAR, Gaver III DP (2005) The pulsatile motion of a semi-infinite bubble in a channel: flow fields, and transport of an inactive surface-associated contaminant. J Fluid Mech 537: 1-33.
- 184. Smith BJ, Gaver III DP (2008) The pulsatile propagation of a finger of air within a fluid-occluded cylindrical tube. J Fluid Mech 601: 1-23. [Crossref]
- 185. Glindmeyer IV HW, Smith BJ, Gaver III DP (2012) In situ enhancement of pulmonary surfactant function using temporary flow reversal. J Appl Physiol 112: 149-158. [Crossref]
- 186. Amin SD, Suki B (2012) Could dynamic ventilation wave forms bring about a paradigm shift in mechanical ventilation? J Appl Physiol 112: 333-334. [Crossref]
- 187. Bohlin K, Gudmundsdottir T, Katz-Salamon M, Jonsson B, Blennow M (2007) Implementation of surfactant treatment during continuous positive airway pressure. J Perinatol 27: 422-427. [Crossref]
- 188. Jing Liu, Sumeng Li, Jia Liu, Boyun Liang, Xiaobei Wang, et al. (2020) Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine: 102763. [Crossref]
- 189. Zhang W, Zhao Y, Zhang F, Wang Q, Li T, et al. (2020) The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The perspectives of clinical immunologists from China. Clin Immunol 214:108393. [Crossref]
- 190. Liao M, Liu Y, Yuan J, Wen Y, Xu G, et al. (2020) The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. MedRxiv: 20026690.
- 191. Fu Y, Cheng Y, Wu Y (2020) Understanding SARS-CoV-2-Mediated `Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol Sin 35: 266-271. [Crossref]
- 192. 192. Pugin J, Verghese G, Widmer MC, Matthay MA (1999) The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med 27: 304-331. [Crossref]
- 193. Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, et al. (2020) Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395: e30-e31. [Crossref]
- 194. Grewal PS, Attia P, McGale JP, Sullivan CM (2020) Exogenous pulmonary surfactant as a potential adjuvant therapy in SARS-CoV-2 patients experiencing acute respiratory distress syndrome? Attia Med PC New York, New York.
- 195. Dantzker D, Wagner P, West JB (1975) Instability of lung units with low ventilation/perfusion ratios during oxygen breathing. J Appl Physiol 38: 886-895.
- 196. Shapiro B, Crane R, Harrison R, Steiner M (1980) Changes in intrapulmonary shunting with administration of 100% oxygen. Chest 77: 138-141.
- 197. Oliver A, Abinader E, Bursztein S (1980) Influence of varying inspired oxygen tensions on intrapulmonary venous admixture (shunt) of mechanically ventilated patients. Crit Care Med 8: 99-101. [Crossref]
- 198. Coetzee A, Swanevelder J, Van der Spuy G, Jansen J (1995) Gas exchange indices – how valid are they? S Afr Med J 85: 1227-1231. [Crossref]
- 199. Karbing DS, Kjærgaard S, Smith BW, Espersen K, Allerød C, et al. (2007) Variation in the PaO2/FiO2 ratio with FiO2: mathematical and experimental description, and clinical relevance. Crit Care 11: R118. [Crossref]
- 200. Blennerhassett JB (1985) Shock lung and diffuse alveolar damage pathological and pathogenetic considerations. Pathology 17: 239-247.[Crossref]
- 201. Idell S (2003) Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med 31: S213-S220. [Crossref]
- 202. Magro CJ, Mulvey J, Berlin D, Nuovo G, Salvatore S, et al. (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res 220: 1-13. [Crossref]
- 203. Marietta M, Ageno W, Artoni A, De Candia E, Gresele P, et al. (2020) The COVID-19 and haemostasis: a position paper from Italian Society on Thrombosis and Haemostasis (SISET) Blood Transfus 18: 167-169. [Crossref]
- 204. Bachofen M, Weibel ER (1982) Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 3: 35-56. [Crossref]
- 205. Hardaway RM, Harke H, Tyroch AH, Williams CH, Vazquez Y, et al. (2001) Treatment of severe acute respiratory distress syndrome: A final report on a phase 1 study. Am Surg 67: 377-382. [Crossref]
- 206. Lu Q, Zhang M, Girardi C, Bouhemad B, Kesecioglu J, et al. (2010) Computed tomography assessment of exogenous surfactant-induced lung reaeration in patients with acute lung injury. Crit Care 14: R135. [Crossref]
- 207. Satoh D, Matsukawa S, Saishu T, Hashimoto Y (1998) Effect of surfactant on respiratory failure associated with thoracic aneurysm surgery. Crit Care Med 26: 1660-1662. [Crossref]
- 208. Hoheisel G, Moche M, Borte G, Bilek K, Schauer J, et al. (1997) Surfactant administration and laterally independent positive pressure ventilation in acute lung failure and atelectasis after septic abortion. Case report. Pneumologie 51: 270-273. [Crossref]
- 209. Staudinger T, Bankier A, Strohmaier W, Weiss K, Locker GJ, et al. (1997) Exogenous surfactant therapy in a patient with adult respiratory distress syndrome after near drowning. Resuscitation 35: 179-182. [Crossref]
- 210. Violi LAD, St John RC, Gadek JE, Pacht ER (1997) Administration of surfactant via flexible bronchoscopy. J Bronchology 4: 329-331.
- 211. Pallua N, Warbanow K, Noaha EM, Machens HG, Poets C, et al. (1998) Intrabronchial surfactant application in cases of inhalation injury: First results from patients with severe burns and ARDS. Burns 24: 197-206. [Crossref]
- 212. Wiswell TE, Smith RM, Katz LB, Mastroianni L, Wong DY, et al. (1999) Cochrane, C.G. 1999. Bronchopulmonary segmental lavage with surfaxin (KL4-surfactant) for acute respiratory distress syndrome. Am J Respir Crit Care Med 160: 1188-1195. [Crossref]
- 213. Krause MF, Von Bismarck P, Oppermann HC, Ankermann T (2008) Bronchoscopic surfactant administration in pediatric patients with persistent lobar atelectasis. Respiration 75: 100-104. [Crossref]
- 214. Krause MF, Ankermann T (2014) Bronchoscopic interventions with surfactant and recombinant human deoxyribonuclease for acute respiratory distress syndrome-type respiratory syncytial virus-pneumonia in moderately preterm infants: Case series. SAGE Open Med Case Rep 2: 2050313X14554479. [Crossref]