Percutaneous coronary intervention (PCI) is the most common form of revascularization for ischemic heart disease. Thrombosis during PCI-which can be fatal-is historically thought to be initiated by a combination of platelet actions and the contact coagulation system. Adjunctive therapy to prevent thrombosis therefore consists of potent antiplatelet and anticoagulant therapies-at the expense of increased, potentially fatal bleeding. However, a fluid dynamic model of coronary flow indicates platelets may slip (i.e.: have non-zero velocity) at device surfaces, functionally extinguishing effects of the contact coagulation system. These findings suggest PCI antithrombotic therapy should focus on antiplatelet and not anticoagulation therapies. We report herein a retrospective analysis of 481 consecutive patients undergoing higher-risk PCI using a strategy of antiplatelet therapy only (oral aspirin, P2Y12 inhibitor; intravenous glycoprotein IIb/IIIa inhibitor), without anticoagulation. The procedural success rate was high (99.2%) with a low rate of 72-hour and 30-day major adverse ischemic events (combined death/myocardial infarction/urgent target vessel revascularization: 2.6% [death-0%] and 4.4% [death-0.8%], respectively). Additionally, bleeding events were low (fatal: 0% and 0.2%, respectively). Thus, PCI can be performed safely and effectively using antiplatelet therapy only, without anticoagulation. If validated, this antiplatelet-focused approach should result in an evidence-based change in the PCI guidelines.
acute coronary syndrome, anticoagulation, antiplatelet drugs, percutaneous coronary intervention, pharmacology, translational science
Percutaneous coronary intervention (PCI) is the most common form of revascularization for ischemic heart disease [1], and is particularly useful for the management of acute coronary syndromes (ACS) [2,3]. Despite >40 years of experience [4], the optimal antithrombotic regimen that concomitantly minimizes both adverse thrombotic and bleeding events has not been determined [5]. Both thrombosis and bleeding significantly increase acute and longer-term morbidity and mortality [6-9]. Moreover, thrombosis has historically been thought by the PCI community to be initiated by a combination of platelet actions and the contact coagulation system [4,10]. Thus, research to identify the optimal regimen has focused on combining drugs that inhibit both specific key receptors of platelet function as well as proteases of the coagulation cascade [5].
It is now understood that, as a rule in the higher-shear environment of PCI, thrombosis occurs in waves, with platelet actions comprising the classic first wave [11,12]. One enduring exception to this rule is activation of the contact coagulation system at PCI device surfaces and consequent thrombosis [13-15]. In the current era of aggressive antiplatelet therapy, this is the sole remaining concern that specifically accounts for the contemporary continued use of anticoagulation during PCI [10,16].
Countering this concern is a fluid dynamic model of coronary flow that indicates platelets may effectively slip (i.e., have non-zero velocity) at PCI device surfaces [17], either naturally and/or under the influence of antiplatelet therapy. In this physical, scaled-up model, which includes a central cylinder representing a PCI guide wire within a cylindrical coronary test section (Figure 1), microspheres-modeling platelets-demonstrated slip immediately adjacent to the central cylinder surface (Figure 1-blue discs). Applying this concept to platelets, their slip would diminish adhesion and functionally extinguish any thrombotic consequence of the contact coagulation system.
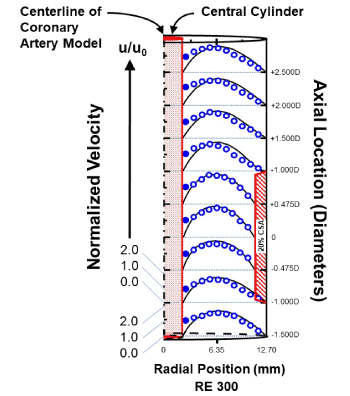
Figure 1. Slip of microspheres in a cylindrical scaled-up model of coronary flow (longitudinal view of horizontal middle plane of hemi-cylinder) [17].
The velocities of 3.12 µm diameter polystyrene microspheres-modelling platelets and as determined by laser-Doppler velocimetry (blue rings and discs: ○;●, respectively)-are compared with COMSOL computer-simulated plasma velocity profiles (black solid lines). The model contains a central solid cylinder representing a PCI guide wire (a “cylinder-within-a-cylinder”), a 20% stenosis by CSA, and the RE of 300 is typical for coronary flow [49]. All velocities are normalized to the mean velocity u0, calculated from direct volumetric measurement of bulk fluid flow. Computer simulations apply the no-slip boundary condition at each surface, as the wall shear stresses in the unobstructed coronary circulation promote minimal plasma slip [73,74]. The blue discs (●) show effective slip (i.e.: non-zero velocity) of microspheres immediately adjacent to the central cylinder surface. (CSA - cross sectional area; PCI - percutaneous coronary intervention; RE - Reynolds number)
These fundamental biologic and fluid dynamic phenomena taken together may explain the success of those very few PCI studies that employed antiplatelet therapy only, without anticoagulation [18-21]. Importantly, those studies involved either low-risk patients, uncomplicated lesions and/or a standard duration infusion of a platelet glycoprotein IIb/IIIa inhibitor (GPI; e.g., eptifibatide: 18-24 hour). Additionally, none of those studies included a description of the local socioeconomic environment, which influences outcomes [22,23].
This report is an extended case series from an exceptionally challenging socioeconomic environment that examines the demographics, key procedural details and outcomes of patients undergoing urgent, higher-risk PCI using antiplatelet therapy only (aspirin, P2Y12 inhibitor and GPI), without the planned use of unfractionated heparin (UFH), bivalirudin or any other anticoagulant. We also sought to determine if there would be tradeoffs with a shortened GPI infusion (≤4.0 hours).
Report Landscape. Southeastern Regional Medical Center (SRMC) is a non-profit hospital located in Robeson County, North Carolina (NC). Geographically, Robeson is the largest of the 100 counties in NC. Characteristics include:
- Rural with a racially and culturally diverse but relatively undereducated population [24]
- 33.1% of the population are in poverty [25]
- Deteriorated from 2nd highest-to-highest age-adjusted death rate from heart disease for the 24 NC counties with established local PCI services during 2011-2017 [26]
- Robert Wood Johnson Foundation County Health Rankings during 2011-2017 demonstrated an annual downward trend from 98th-to-100th place out of the 100 NC counties [27].
The cardiac catheterization laboratory, operated during the report period by Duke University Medical Center (DUMC) faculty, had access to only basic PCI devices (e.g., early second-generation drug-eluting coronary stents). Additionally, it had no systematic quality assurance program. In an effort to quantitate outcomes and identify areas for improvement, an automated quality assurance system was initiated. One operator, a longstanding proponent of an antiplatelet-focused strategy in PCI (SJD [28]), created a comprehensive database to be used for internal validation of the automated system, and to promote process improvement for all PCI procedures performed at SRMC. Design and conduct of the analysis herein were approved by both SRMC and DUMC Institutional Review Boards.
The antiplatelet-focused strategy used in the patients described in this analysis was adopted by the operator [SJD] in March, 1997, for the sole intention of optimizing PCI outcomes within the scope of contemporary clinical practice [29,30]. This date was 4 years before guidelines were published that included recommendations for anticoagulant dosing during PCI [31]. Subsequent iterations in the strategy were relatively minor and included, beginning in January 2012, progressively briefer infusions of GPI (ultimately to ≤4.0 hours). Continued surveillance of the outcomes of these patients remained consistently superior to the results of contemporary published reports [18,21,28]. As a consequence, per the Belmont Report [32], the use of the antiplatelet-focused strategy described in this report did not constitute research or experimentation. Instead, this report describes a consistent variance within scope of practice. All methods were therefore carried out in accordance with relevant guidelines and regulations.
Patient Population: The database consisted of all patients undergoing PCI from December, 2011, through May, 2017 (Figure 2). The only patients transferred from SRMC to DUMC for PCI were those requiring PCI ancillary devices not available at SRMC (e.g., rotational atherectomy, advanced mechanical circulatory support). Inclusion criteria for this report consisted of a formal diagnosis of acute, non-ST segment elevation myocardial infarction (NSTEMI), unstable angina (USA), or acute ischemic congestive heart failure (CHF) requiring intravenous diuretic therapy in the absence of NSTEMI or USA. Exclusion criteria included (Figure 2): (1) pre-PCI STEMI and/or cardiac arrest ≤24 hours; (2) treatment with enoxaparin ≤8 hours or UFH ≤2 hours pre-PCI; (3) lack of treatment with dual oral antiplatelet therapy (DAPT) pre-PCI; (4) PCI without stent deployment; (5) PCI of a bypass graft; and (6) patient a legal ward of the state. All patients provided informed written consent prior to PCI.
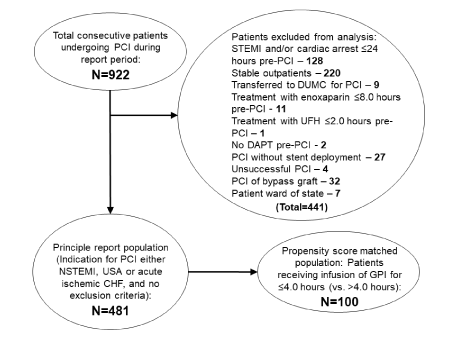
Figure 2. Flow diagram for the report.
Of the 922 patients undergoing PCI during the report period, 481 patients were eligible for this report (with 441 patients excluded). There were 4 unsuccessful PCIs among the excluded patients: 3 involved chronic total occlusions; the other involved a target lesion within a large side branch of a major coronary artery, with the side branch behind 2 layers of stent previously deployed within the parent coronary artery (“double stent jail”). None of the 4 unsuccessful PCI patients experienced a 72-hour or 30-day adverse event. (CHF - congestive heart failure; DAPT – oral dual antiplatelet therapy; NSTEMI - non-ST segment elevation myocardial infarction; PCI - percutaneous coronary intervention; UFH - unfractionated heparin; USA - unstable angina)
Antithrombotic therapy: Each patient was maintained on oral aspirin (81-325 mg daily) and, once the decision made to proceed to PCI, loaded with an oral P2Y12 inhibitor (if not already at steady state; loading dose: ticagrelor-180 mg; clopidogrel-600 mg; prasugrel-60 mg). Also, each patient received an infusion of a GPI (eptifibatide with double bolus: for normal renal function, 180 µg/kg bolus followed by a continuous infusion at 2 µg/kg/min, with another 180 µg/kg bolus 10 minutes after initial bolus; for non-hemodialysis-dependent impaired renal function, same but continuous infusion reduced to 1 µgm/kg/min; hemodialysis-dependent patients [N=6] received abciximab: 0.25 mg/kg bolus followed by a continuous infusion at 0.125 μg/kg/min [to a maximum of 10 μg/min]). The infusion duration was ≤18.0 hours (eptifibatide) or ≤12.0 hours (abciximab), and was defined from the time of administration of the initial bolus to nursing documentation of discontinuation. The only exposure to an anticoagulant during PCI was UFH contained within the flush solution (1000 Units/500 mL, and with a typical net volume administered by completion of PCI of 50-75 mL).
PCI technique and angiographic assessment: All PCI procedures were performed using 6 French guiding catheters with fluoroscopic-guided femoral artery access, and each PCI was initiated following completion of the bolus infusion(s) of GPI. Also, each patient received intravenous fentanyl to facilitate sedation. Finally, angiograms were assessed using quantitative methods for both morphology and flow [33].
Post-PCI management: All sheaths were removed immediately post-PCI, following confirmation of an activated clotting time <170 seconds (ACT Plus Automated Coagulation Timer; Medtronic; Minneapolis, MN; normal range, 90-130 sec). Manual compression was applied followed by 6 hours of bedrest. An electrocardiogram was performed upon PCI completion. Additionally, at 12-18 hours post-PCI, a repeat electrocardiogram, measurements of serum troponin I, complete blood count and chemistry panel were acquired. All patients were assessed at the bedside on the day following PCI, and again ≥30 days post-PCI.
Outcome endpoints: Major adverse cardiac events (MACE) were defined as all-cause death, MI [34] and target vessel revascularization. Type 4a MI was defined as a post-PCI increase in troponin to >5x the 99th percentile upper reference limit (URL) or to >20% above the previous nadir with clinical, electrocardiographic, angiographic or imaging-positive ischemic findings. Type 4b MI was defined by angiographic-documented stent thrombosis. Intraprocedural thrombotic events (IPTE) were defined as new or increasing thrombus, abrupt vessel closure, no-/slow-reflow, or ST-segment deviation in the absence of the preceding criteria and of >60 seconds duration following balloon deflation. Bleeding Academic Research Consortium (BARC) criteria were used to classify bleeding events [9]. Outcome endpoints were tabulated at 72 hours and 30 days.
Statistical Analysis: Continuous variables are reported as the mean ± standard deviation. Categorical variables are reported as number (percent) for each GPI treatment group (short duration: ≤4.0; vs. more standard duration: >4.0-18.0 hours). Continuous variables were tested for normality using the Kolmogorov-Smirnov test. Subsequent analysis involved the two-sample t-test if found normal or the Wilcoxon rank sum test if not normal. Categorical data were analyzed with the chi-squared or Fisher’s exact test, depending upon cell count.
Propensity score matching of short and more standard duration GPI infusion groups employed a logistic regression model based on demographic and clinical characteristics prior to PCI (Tables 1 and 2, footnotes). Propensity matching was 1:1 with a maximal caliper width of 0.15; thus all 100 short-infusion patients were matched to 100 more standard duration patients. The standardized mean differences (SMD) for both the unmatched and matched groups were calculated with the goal of minimizing the SMD for each variable. The relationship between GPI infusion groups and each endpoint was then assessed using a repeated measures logistic regression model to account for the correlation of matched pairs. Results are presented as the odds ratio (OR) with 95% confidence interval (CI).
Outcome endpoints were also uniformly low at 30 days (Table 4), including 4.4% MACE (death-0.8%). There was only one (0.2%) BARC 5 bleeding event (fatal gastrointestinal hemorrhage on day 27 post-PCI). The BARC (2+3) bleeding event rate was 3.3% (BARC 2: 1.2%; BARC 3: 2.1%).
Table 1. Baseline characteristics of 481 report patients based upon GPI infusion duration, both before and after matching*
Baseline Characteristics |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value
(Unmatched Groups)
[SMD] |
P-value
(Matched Groups)
[SMD] |
GPI Duration:
(Sample Size) |
Age (years) |
61.9±11.7 |
62.9±11.4 |
62.4±12.6 |
0.61 [0.04] |
0.78 [0.04] |
Male sex |
226 (59.3%) |
59 (59.0%) |
54 (54.0%) |
0.34 [0.11] |
0.56 [0.10] |
Race |
|
|
|
0.30 [0.12] |
0.80 [0.05] |
Caucasian |
181 (47.5%) |
52 (52.0%) |
50 (50.0%) |
|
|
African-American |
69 (18.1%) |
25 (25.0%) |
23 (23.0%) |
|
|
Native-American |
131 (34.4%) |
23 (23.0%) |
27 (27.0%) |
|
|
Married† |
214 (56.2%) |
57 (57.0%) |
49 (49.0%) |
0.20 [0.15] |
0.30 [0.16] |
Insurance type† |
|
|
|
0.56 [0.30] |
0.60 [0.35] |
Non-private‡ |
258 (67.7%) |
69 (69.0%) |
62 (62.0%) |
|
|
Private |
80 (21.0%) |
21 (21.0%) |
25 (25.0%) |
|
|
Uninsured |
43 (11.3%) |
10 (10.0%) |
13 (13.0%) |
|
|
BMI (kg/m2) |
31.0±7.8 |
30.0±6.8) |
30.1±7.3 |
0.34 [0.12] |
0.84 [0.014] |
Major CHD risk factors: |
|
|
|
|
|
Diabetes mellitus |
181 (47.5%) |
42 (42.0%) |
51 (51.0%) |
-- |
-- |
Insulin-dependent |
89 (23.4%) |
19 (19.0%) |
23 (23.0%) |
0.72 [0.10] |
0.45 [0.13] |
Non-insulin-dependent |
92 (24.1%) |
23 (23.0%) |
28 (28.0%) |
Hypertension |
346 (90.8%) |
86 (86.0%) |
83 (83.0%) |
0.03 [0.23] |
0.71 [0.08] |
Dyslipidemia |
365 (95.8%) |
92 (92.0%) |
93 (93.0%) |
0.29 [0.12] |
1.00 [0.04] |
Cigarette smoking-active |
137 (36.0%) |
34 (34.0%) |
33 (33.0%) |
0.67 [0.06] |
0.99 [0.04] |
Family history of premature CHD† |
346 (91.1%) |
91 (91.0%) |
91 (91.0%) |
0.99 [<0.01] |
1.00 [<0.01] |
Other co-morbidities |
|
|
|
|
|
Prior MI |
161 (42.3%) |
33 (33.0%) |
35 (35.0%) |
0.19 [0.15] |
0.88 [0.04] |
Prior revascularization§ |
223 (58.5%) |
50 (50.0%) |
52 (52.0%) |
0.24 [0.13] |
0.88 [0.04] |
Cerebral vascular disease |
53 (13.9%) |
22 (22.0%) |
21 (21.0%) |
0.08 [0.19] |
1.00 [0.02] |
Peripheral vascular disease |
42 (11.0%) |
9 (9.0%) |
10 (10.0%) |
0.77 [0.03] |
1.00 [0.03] |
Chronic obstructive pulmonary disease |
141 (37.0%) |
30 (30.0%) |
31 (31.0%) |
0.27 [0.13] |
1.00 [0.02] |
Chronic renal insufficiency |
93 (24.4%) |
15 (15.0%) |
13 (13.0%) |
0.01 [0.30] |
0.83 [0.06] |
Clinical presentation: |
|
|
|
0.26 [0.12] |
0.97 [0.04] |
NSTEMI |
177 (46.5%) |
50 (50.0%) |
51 (51.0%) |
|
|
USA |
155 (40.7%) |
42 (42.0%) |
42 (42.0%) |
|
|
CHF |
49 (12.9%) |
8 (8.0%) |
7 (7.0%) |
|
|
Heart failure class at admission (NYHA) |
|
|
|
<0.01 [0.84] |
0.84 [0.19] |
1 or 2 |
133 (34.9%) |
24 (24.0%) |
21 (21.0%) |
|
|
3 |
162 (42.5%) |
58 (58.0%) |
62 (62.0%) |
|
|
4 |
86 (22.6%) |
18 (18.0%) |
17 (17.0%) |
|
|
Left ventricular ejection fraction (%) |
43.0±9.0 |
43.8±8.1 |
43.7±7.1 |
0.91 [0.09] |
0.96 [0.01] |
BMI=Body mass index, CHD=Coronary heart disease, CHF=Congestive Heart Failure (acute, and requiring intravenous diuretic therapy),
GPI=Glycoprotein inhibitor, NSTEMI=Non-ST Segment Elevation Myocardial Infarction, NYHA=New York Heart Association,
SMD=Standardized Mean Difference, USA=Unstable Angina.
*Data are presented as mean ± standard deviation or as number (percentage).
†Variable not used in propensity score matching.
‡Non-private insurance: Medicare, Medicaid or Veterans Affairs.
§Prior revascularization: Percutaneous or surgical.
P-value (Unmatched Groups): Comparison between GPI ≤4.0 hours (N=100) vs.GPI >4.0 hours-unmatched group (N=381).
P-value (Matched Groups): Comparison between GPI ≤4.0 hours (N=100) vs.GPI >4.0 hours matched group (N=100).
Table 2. PCI details based upon GPI infusion duration, both before and after matching*
PCI Details |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value
(Unmatched Groups)
[SMD] |
P-value
(Matched Groups)
[SMD] |
GPI Duration:
(Sample Size) |
GPI duration (hour)† |
11.2±4.0 |
11.2±6.8 |
3.6±0.6 |
<0.01 [2.66] |
<0.01 [1.57] |
Activated clotting time (sec.) |
145.4±16.7 |
144.7±16.7 |
142.4±18.2 |
0.21 [0.17] |
0.39 [0.13] |
Pre-PCI anticoagulation |
|
|
|
|
|
Unfractionated heparin† |
137 (36.0%) |
33 (33.0%) |
38 (38.0%) |
0.71 [0.04] |
0.52 [0.11] |
Enoxaparin |
164 (43.0%) |
48 (48.0%) |
43 (43.0%) |
0.96 [0.01] |
0.55 [0.10] |
Final Dose (mg/kg) |
0.75±0.27 |
0.74±0.27 |
0.64±0.27 |
0.02 [0.41] |
0.15 [0.37] |
Time of final dose pre-PCI (hour)† |
18.1±8.7 |
18.1±7.7 |
17.9±8.2 |
0.97 [0.02] |
0.38 [0.03] |
No anticoagulation† |
80 (21.0%) |
19 (19.0%) |
19 (19.0%) |
0.66 [0.05] |
1.00 [<0.01] |
Pre-PCI P2Y12 inhibitor‡ |
|
|
|
|
|
Already receiving pre-PCI |
263 (69.0%) |
51 (51.0%) |
53 (53.0%) |
0.03 [0.33] |
0.88 [0.04] |
Loaded at time of PCI |
164 (43.0%) |
79 (79.0%) |
79 (79.0%) |
<0.01 [0.79] |
1.00 [<0.01] |
Lesion characteristics: |
|
|
|
|
|
Multi-vessel |
105 (27.6%) |
21 (21.0%) |
23 (23.0%) |
0.36 [0.11] |
0.87 [0.05] |
De novo |
330 (86.6%) |
92 (92.0%) |
88 (88.0%) |
0.72 [0.04] |
0.48 [0.13] |
Lesion type B2 or C |
206 (54.1%) |
60 (60.0%) |
60 (60.0%) |
0.54 [0.26] |
0.95 [0.07] |
Stenosis severity
(% by diameter): |
|
|
|
|
|
Pre-PCI |
90.5±9.2 |
90.1±9.9 |
90.5±9.6 |
0.83 [<0.01] |
0.80 [0.04] |
Post-PCI |
-2.3±6.3 |
-1.3±5.9 |
-1.2±6.3 |
0.07 [0.18] |
0.95 [0.02] |
TIMI flow: |
|
|
|
|
|
Pre-PCI |
2.45±0.86 |
2.35±0.95 |
2.35±0.88 |
0.18 [0.12] |
1.00 [<0.01] |
Post-PCI |
2.96±0.20 |
2.94±0.24 |
2.95±0.22 |
0.73 [0.05] |
0.76 [0.04] |
Stent design: |
|
|
|
|
|
Drug-eluting |
213 (55.9%) |
41 (41.0%) |
43 (43.0%) |
0.02 [0.26] |
0.89 [0.04] |
Diameter (mm) |
2.56±0.45 |
2.53±0.45 |
2.53±0.51 |
0.23 [0.06] |
0.94 [<0.01] |
Length (mm) |
21.3±11.4 |
19.5±8.1 |
19.4±7.9 |
0.46 [0.19] |
0.89 [0.02] |
Maximum deployment pressure (atm) |
17.2±2.7 |
16.9±2.7 |
16.8±3.3 |
0.10 [0.13] |
0.86 [0.03] |
Procedure duration (min.)§ |
29.6±15.2 |
26.1±12.0 |
28.0±19.5 |
0.15 [0.09] |
0.44 [0.12] |
Length of stay post-PCI (days)†
Length of stay range (days)† |
1.5±1.4
1-14 |
1.5±1.7
1-14 |
1.7±3.0
1-27 |
0.48 [0.09] |
0.76 [0.08] |
GPI=Glycoprotein inhibitor, PCI=Percutaneous coronary intervention.
*Data are presented as mean ± standard deviation or as number (percentage).
†Variable not used for propensity score matching.
‡Pre-PCI P2Y12 inhibitor: If unclear whether patient fully loaded pre-PCI, then loading dose administered at that time.
§Procedure duration: Defined from the time of insertion of 1st guiding catheter (if multiple) to acquisition of final angiographic images.
P-value (Unmatched Groups): Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-unmatched group (N=381).
P-value (Matched Groups): Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-matched group (N=100).
All tests were two-sided; a p-value <0.05 was considered statistically significant. Statistical analyses were performed using SAS v9.4 (SAS Institute, Inc., Cary, NC) by the Duke Department of Biostatistics and Bioinformatics (Durham, NC).
During the report period, 922 patients underwent PCI; 481 were eligible for this report (Figure 2). The most common reason for exclusion was a presentation with STEMI, cardiac arrest or stable outpatient (N=348). Transfer to DUMC for PCI, treatment with enoxaparin or UFH within defined time frames and no pre-PCI DAPT were rare (N=9, 12 and 2, respectively).
Baseline characteristics of the reported patients included a high prevalence of major cardiovascular disease risk factors (Table 1; including diabetes mellitus-48.2%) and persisted in the matched cohort. The most common individual clinical presentation was a NSTEMI (47.4%) with a decrease in left ventricular ejection fraction to below the normal range (i.e.: <55%).
The short GPI infusion patient cohort was more likely to receive a lower final dose of enoxaparin pre-PCI, more likely to receive P2Y12 inhibitor loading immediately pre-PCI, and less likely to receive a drug-eluting stent (Table 2). All other PCI details were similar both before and, including these three details, after matching. Other higher-risk characteristics included a disproportionately high percentage of patients with type B2/C target lesions, multivessel PCI, and smaller target vessel diameter [18-21, 35-40].
The procedure success rate was 99.2%. Outcome endpoints were uniformly low at 72 hours (Table 3), including 2.6% MACE (all type 4a MI; death-0%). There were no BARC 5 bleeding events. The BARC (2+3) bleeding event rate was 2.1% (BARC 2: 0.8%; BARC 3: 1.3%). During PCI, IPTE occurred with a similar frequency both before and after matching for GPI duration (overall incidence, 11.2%), consisting solely of transient slow reflow and/or ST-segment deviation. There was no angiographically-documented thrombosis or 72-hour type 4b MI. However, IPTE was associated with subsequent 72-hour type 4a MI (p<0.001). All type 4a MIs were uncomplicated, required no specific new intervention and did not prolong hospitalization.
Table 3. 72-hour outcomes based upon GPI infusion duration, both before and after matching*
Outcomes |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value |
OR (CI) |
GPI Duration:
(Sample Size) |
MACE |
11 (3.0%) |
5 (5.2%) |
1 (1.1%) |
0.22 |
0.20 (0.00-1.79) |
Death |
0 |
0 |
0 |
-- |
-- |
MI† |
11 (3.0%) |
5 (5.2%) |
1 (1.1%) |
0.22 |
0.20 (0.00-1.79) |
Q-wave |
0 |
0 |
0 |
-- |
-- |
Type 4a MI |
11 (3.0%) |
5 (5.2%) |
1 (1.1%) |
0.22 |
0.20 (0.00-1.79) |
Type 4b MI |
0 |
0 |
0 |
-- |
-- |
TVR |
0 |
0 |
0 |
-- |
-- |
TLR |
0 |
0 |
0 |
-- |
-- |
IPTE |
46 (12.1%) |
13 (13.0%) |
8 (8.0%) |
0.38 |
1.63 (0.62-4.52) |
Subsequent MACE |
9 (19.6%) |
4 (30.8%) |
1 (12.5%) |
-- |
-- |
Bleeding events |
|
|
|
|
|
BARC 2 |
4 (1.0%) |
1 (1.0%) |
0 |
|
1.00 (0.00-19.00) |
BARC 3‡ |
5 (1.3%) |
3 (3.0%) |
1 (1.0%) |
|
0.33 (0.01-4.15) |
BARC 5 |
0 |
0 |
0 |
-- |
-- |
Thrombocytopenia |
6 (1.6%) |
2 (2.0%) |
0 |
0.25 |
0.41 (0.00-3.47) |
Profound (<20,000/μl) |
0 |
0 |
0 |
-- |
-- |
BARC=Bleeding Academic Research Consortium9, BARC 2=Any overt, actionable sign of bleeding that does not fit the criteria for BARC type 3-5, BARC 3=Overt bleeding plus hemoglobin drop of 3 to <5 g/dL (provided hemoglobin drop is related to bleed); any transfusion with overt bleeding, BARC 5= Fatal bleeding.
GPI=Glycoprotein inhibitor, IPTE=Intraprocedural thrombotic events, MACE=Major adverse cardiac events (death from any cause; Type 4 myocardial infarction; target vessel revascularization), MI=Myocardial infarction, TLR=Target lesion revascularization (including both urgent and non-urgent), TVR=Target vessel revascularization.
*Data are presented as number (percentage).
†N=21 patients excluded from 72-hour analysis due to pre-PCI increasing troponin I (Unmatched N excluded=16/381; Matched GPI >4.0 hour N excluded=4/100; GPI ≤4.0 hour N excluded=5/100).
‡BARC 3 bleeding events: All type 3a.
P-value: Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-matched group (N=100).
Outcomes not mutually exclusive.
Table 4. 30-Day outcomes based upon GPI infusion duration, both before and after matching*
Outcomes |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value |
OR (CI) |
GPI Duration:
(Sample Size) |
MACE |
18 (4.7%) |
5 (5.0%) |
3 (3.0%) |
0.73 |
0.60 (0.09-3.08) |
Death |
3 (0.8%) |
0 |
1 (1.0%)† |
0.50 |
1.00 (0.05-ꝏ) |
MI‡ |
|
|
|
|
|
Type 1§+4a |
14 (3.7%) |
5 (5.0%) |
2 (2.0%) |
0.45 |
0.40 (0.04-2.44) |
Type 4b |
2 (0.5%)¶ |
0 |
0 |
-- |
-- |
TVR |
3 (0.8%) |
0 |
1 (1.0%) |
0.50 |
1.00 (0.05-ꝏ) |
TLR |
2 (0.5%) |
0 |
0 |
-- |
-- |
Bleeding events |
|
|
|
|
(compared to 0) |
BARC 2 |
6 (1.6%) |
1 (1.0%) |
0 |
0.22 |
1.00 (0.00-19.00) |
BARC 3 |
9 (2.4%) |
5 (5.0%) |
1 (1.0%) |
0.20 (0.00-1.79) |
BARC 5 |
0 |
0 |
1 (1.0%)† |
1.00 (0.05-ꝏ) |
Readmission |
72 (18.9%) |
17 (17.0%) |
12 (12.0%) |
0.41 |
0.64 (0.25-1.60) |
Principle diagnosis:
Cardiovascular |
17 (23.6%) |
5 (29.4%) |
3 (25.0%) |
0.50 |
1.00 (0.00-19.00) |
Principle diagnosis:
Non-cardiovascular |
55 (76.4%) |
12 (70.6%) |
9 (75.0%) |
BARC=Bleeding Academic Research Consortium9, BARC 2=Any overt, actionable sign of bleeding that does not fit the criteria for BARC type 3-5,
BARC 3=Overt bleeding plus hemoglobin drop of 3 to <5 g/dL (provided hemoglobin drop is related to bleed); any transfusion with overt bleeding,
BARC 5= Fatal bleeding.
GPI=Glycoprotein inhibitor, IPTE=Intraprocedural thrombotic events, MACE=Major adverse cardiac events (death from any cause; myocardial infarction; target vessel revascularization). MI=Myocardial infarction, TLR=Target lesion revascularization (including both urgent and non-urgent), TVR=Target vessel revascularization.
*Data are presented as number (percentage).
†Death on day 27 post-PCI: Documented fatal gastrointestinal bleed (Non-IPTE patient).
‡N=21 patients excluded from 72-hour analysis due to pre-PCI increasing troponin I (Unmatched N excluded=16/381; Matched GPI >4.0 hour N excluded=4/100; GPI ≤4.0 hour N excluded=5/100).
§Each NSTEMI.
¶Both events involved same Non-IPTE patient with documented medical non-compliance.
P-value: Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-matched group (N=100).
Outcomes not mutually exclusive.
Finally, after matching, MACE and BARC (2+3) bleeding events were both numerically less frequent in the short-infusion group at 72 hours and 30 days compared with the matched group (Table 4; differences not statistically significant). During the 30 days post-PCI, 1 death occurred within the short-infusion group, resulting from the sole BARC 5 bleeding event. All remaining event rates were low and readmission rates similar, both before and after matching.
This report shows that higher-risk PCI can be performed safely and effectively using antiplatelet therapy only (aspirin, P2Y12 antagonist, and GPI), without adjunctive parenteral anticoagulation. Empirically, few clinical events were observed, with rates of both MACE and bleeding being comparable or lower than those reported in the contemporary literature [1,35-41]. Additionally, a shorter duration GPI infusion resulted in a numeric reduction in BARC (2+3) bleeding events at 72 hours and 30 days, without a trade-off in ischemic complications. Finally, this report places these salutary outcomes in the context of: (1) an exceptionally challenging socioeconomic environment; and (2) a patient population with a very high prevalence of major cardiovascular disease risk factors (including diabetes mellitus-48.2%). This environment and patient population increased the risk for PCI-associated adverse events including MACE and bleeding [7,8,22,23,42], and therefore amplify the positive outcomes we report.
The antithrombotic regimen described in this report–specifically, antiplatelet therapy only, without parenteral anticoagulation-is based upon predicate research delineating the mechanisms of thrombus formation in a high-shear environment and informed by studies of fluid dynamics. The predicate research has shown that, at a site of vascular injury and particularly at high shear, the first wave of thrombosis involves a shear-induced conformational change in plasma von Willebrand factor (vWf), producing activation (vWfa), which then activates platelets via the shear receptor complex, GPIb-IX-V [11,12,43,44]. In this environment it is the second wave of thrombosis that is mediated by proteases of the coagulation cascade [11,12].
Predicate research has also shown that contact by coagulation protease factor XII (Hageman) with device surfaces results in its adsorption and a consequent change in its conformation producing activation (factor XIIa; Figure 3A) [45]. Factor XIIa then initiates a series of reactions involving factor XI (plasma thromboplastin), factor IX (Christmas), plasma prekallikrein and factor XII itself (via reciprocal activation by plasma kallikrein). This contact coagulation system-or “intrinsic” pathway-catalyzes the “extrinsic” and final common pathways of coagulation which ultimately convert factor II (thrombinogen) into factor IIa (thrombin). As a principal function, factor IIa hydrolyses factor I (fibrinogen) into factor Ia (fibrin-the fine meshwork that, with factor XIII, provides a superstructure for aggregated platelets in the formation of thrombus). Finally, independent of the contact coagulation system, naïve platelets can directly adhere to device surfaces through inactivated platelet glycoprotein IIb/IIIa binding to surface-adsorbed factor I and other proteins [45], followed by their activation. The adhesion to device surfaces is amplified for platelets already in the activated state.
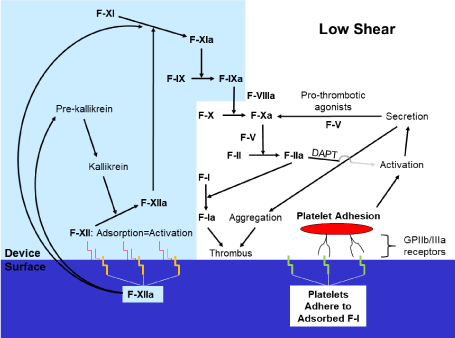
Figure 3. Activity of the coagulation cascade and platelets at a PCI device surface.
The contact coagulation system (light blue shade) is initiated by contact and adsorption of factor XII (Hageman) onto device surfaces with a consequent change in its conformation producing activation (factor XIIa) [45]. Other proteins, including factor I (fibrinogen), are adsorbed by device surfaces as well. Moreover, further down the coagulation cascade, the effect of factor IIa (thrombin) on platelet activation during PCI performed in the context of NSTE-ACS is diminished by DAPT [46,47].
Panel A: In a low-shear environment, naïve platelets can adhere via the inactivated GP IIb/IIIa receptor to adsorbed factor I (and other proteins) and then become activated, secrete and aggregate. This mechanism is amplified for activated platelets. However, in a low-shear environment, the contact coagulation system dominates [48]. Platelet aggregates, in conjunction with the end-product of the coagulation cascade, factor Ia (fibrin), form thrombus.
DAPT=dual oral antiplatelet therapy; F=Coagulation factor; GP=Glycoprotein; NSTE-ACS=Non-ST segment elevation acute coronary syndrome; PCI=Percutaneous coronary intervention; vWfa=activated von Willebrand factor.
As a secondary function, factor IIa is a potent activator of platelets via the protease activated receptor (PAR)-1 and PAR-4. [44]. However, multiple studies have shown strong inhibitory effects of P2Y12 inhibitors on PAR-1 and PAR-4-mediated platelet activation and aggregation, both in platelet-rich plasma and in whole blood [46] (Figure 3A, 3B). Additionally, for PCI employing DAPT and performed in the context of NSTE-ACS, adjunctive inhibition of PAR-1 (using vorapaxar) adds no significant incremental benefit in preventing MACE [47].
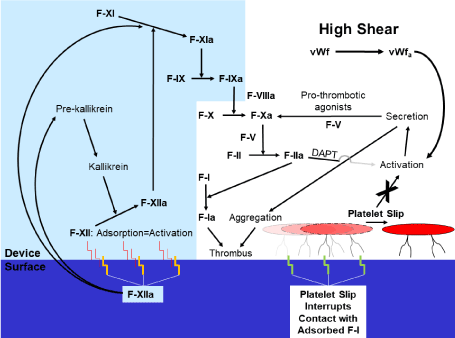
Figure 3. Activity of the coagulation cascade and platelets at a PCI device surface.
Panel B: In a high-shear environment, platelet slip at the device surface interrupts physical contact between the platelet and that surface, therefore diminishing adhesion and functionally extinguishing effects of the contact coagulation system. Thus, no thrombus forms by this mechanism. However, high shear can nonetheless activate platelets via vWfa [11,12,43,44], with ultimate thrombus formation.
DAPT=dual oral antiplatelet therapy; F=Coagulation factor; GP=Glycoprotein; NSTE-ACS=Non-ST segment elevation acute coronary syndrome; PCI=Percutaneous coronary intervention; vWfa=activated von Willebrand factor.
Importantly, in-vivo the contact coagulation system dominates thrombosis at low shear rates (<50 sec-1; resulting in erythrocyte-rich, “red” thrombus) while platelet actions dominate at high shear rates (>5000 sec-1; resulting in platelet-rich, “white” thrombus) [48]. Shear rates in native coronary arteries without atherosclerotic narrowing are in the intermediate-to-high range (700-800 sec-1) [49]. The presence of obstructive atherosclerotic disease increases shear rates further [50], thus favoring platelet-mediated thrombosis [43,44].
Our studies of fluid dynamics, which were conducted in a higher-shear environment similar to the coronary circulation, indicate platelets may demonstrate a shear-dependent slip at PCI device surfaces (Figure 1) [17]. Platelet slip would interrupt physical contact between the platelet and that surface, therefore diminishing adhesion and functionally extinguishing effects of the contact coagulation system (and surface-adsorbed factor I) (Figure 3B). However, platelet slip would not be anticipated in studies of PCI devices conducted in a static or low-shear environment (and where the contact coagulation system dominates) [14,15].
Despite slip, the introduction of PCI devices into the arterial system can nonetheless directly promote thrombosis adjacent to their surfaces by two shear/platelet-associated mechanisms. First, high plasma shear rates are generated adjacent to device and vessel surfaces [51,52] (Figure 1-black solid lines [17]). These high shear rates promote platelet-mediated thrombosis (via vWfa-Figure 3B) with adhesion to local device surfaces that is refractory to therapeutic UFH [53]. Second, superimposed disordering of platelet shear rates distal to stenoses along PCI device surfaces [17] can activate platelets via multiple mechanisms independent of vWf [54]. Thus, while platelet slip may render anticoagulation unnecessary, prevention of thrombosis on PCI devices in the arterial system does mandate antiplatelet therapy.
In addition to high shear, two platelet activation pathways of particular importance in the first wave of thrombosis involve thromboxane A2 and adenosine diphosphate [11,12,44,46]. Clinically, DAPT targeting the associated cyclooxygenase (COX)-1 and P2Y12 platelet receptors diminishes platelet activation to the point that patients presenting with NSTE-ACS can be stabilized without anticoagulation [55] (and the addition of vorapaxar adds no incremental benefit in preventing PCI-associated MACE [47]). Also, low-risk PCI can be performed safely and effectively using DAPT, without anticoagulation (nor GPI) [20]. Thus, for higher-risk PCI, antiplatelet therapy only, using (COX)-1 and P2Y12 platelet receptor inhibitors with, as a safety net, a GPI (blocking glycoprotein IIb/IIIa binding to surface-adsorbed factor I, other surface proteins as well as the final common pathway of platelet aggregation) but no anticoagulation should be safe and effective [28].
Indeed, the safety and efficacy of this strategy are shown by our reported clinical outcomes. In particular, the low incidences of 72-hour and 30-day MACE in this report (Tables 3 and 4) are similar to incidences cited in those very few PCI studies that employed antiplatelet therapy only for treatment in either low-risk patients, uncomplicated lesions and/or a standard duration GPI infusion [18-21]. Additionally, bleeding event rates appear similarly low. The low incidences of MACE in this report also compare favorably with contemporary reports of similar patient populations who received anticoagulation during PCI. For example, the most recent American Heart Association statistic for in-hospital death among NSTEMI patients who undergo PCI is 1.45% [1]. Also, the incidence of in-hospital and 30-day MACE have been reported at 4.0-7.5% and 7.5-14.1%, respectively [35-38, 41].
To provide context for the rate of 72-hour type 4a MI for our entire cohort (2.6%), 19.8-44.2% of NSTE-ACS patients undergoing PCI demonstrate a new troponin elevation >5x the 99th percentile URL or >20% above the previous nadir [39,56]. Thus, our results are favorable and, importantly, parallel the decrease in ischemic complications documented in the group randomized to no-UFH vs. therapeutic-UFH in the Coronary Interventions Antiplatelet-based Only (CIAO) Study [20] (low-risk PCI with steady state DAPT). This parallel underscores the paradoxical platelet activating and other pro-thrombotic potentials of UFH [57-60].
IPTE occurred in 11.2% of our entire cohort, which is within the published range for urgent PCI (3.5-11.4% [38,40]). However, our IPTE were confined to transient slow reflow and/or ST-segment deviation. Remarkably, no IPTE patient experienced a 72-hour/30-day type 4b MI or BARC 5 bleeding event (compared with 3.3%/5.8% and 12.4%/12.4%, respectively [38]).
The zero rate of BARC 5 and the low rates of BARC (2+3) bleeding at 72 hours (2.1%) compete with the exemplary 48-hour rates reported in the Cangrelor versus Standard Therapy to Achieve Optimal Management of Platelet Inhibition (CHAMPION) PCI trial (Thrombolysis in Myocardial Infarction [TIMI] major: 0.4%; minor+access-site bleeding requiring radiologic or surgical intervention: 0.9%) [35] and compare favorably with rates reported for other similar populations receiving anticoagulation (with ranges of in-hospital and 30-day major/minor bleeding: 1.4-10.2%/3.4-8.0% [8,38,41,61-63] and 1.7-10.2%/4.0-8.0% [8,36-38,41,62,63]). Our low rates of bleeding events are consistent with UFH’s dose-dependent relationship with bleeding [59,64,65].
Finally, this report provides a foundation to: (1) extend previous research seeking the lowest effective dose of UFH in higher-risk PCI when performed with adequate antiplatelet therapy [65] (evidently zero); (2) avoid early rebound ischemic events in patients receiving anticoagulation for treatment of their presenting ACS [66]; (3) avoid inadequate oral P2Y12 inhibition at the time of PCI due to the time required for drug absorption, which can be prolonged by fentanyl and other opioids [67]; (4) avoid use of an FDA-approved drug (UFH) for a non-FDA-approved indication (PCI) [68]; (5) support same-day discharge for morning PCI performed via femoral artery access and with a short duration GPI infusion [69]; (6) re-affirm femoral artery access for PCI-thus avoiding radial artery access, its attendant complications [70], and therefore preserving the radial artery as conduit for future use in bypass grafting [71]; (7) enjoy significant cost-savings resulting from the decrease in ischemic and bleeding complications (estimated annual savings for similar PCI patients in United States during index hospitalization: $200-500M) [72]; and (8) highlight translation of basic biologic science and experimental fluid dynamics into clinical application of therapeutics.
This report has several limitations. First, it was a retrospective analysis of a series of patients managed by a single institution. Second, despite discontinuation of anticoagulation therapy pre-PCI, a sub-therapeutic effect may have nonetheless been present during PCI (due to residual effects and the low doses of UFH in the flush solution). Third, selection bias may have occurred when deciding the GPI infusion duration. Finally, ascertainment errors may have been introduced by the lack of scheduled measurements of troponin I timed relative to the PCI procedure itself.
This report challenges the conventional wisdom of the need for anticoagulation as an adjunct to PCI. A combination of aspirin, P2Y12 inhibitor, and short infusion duration GPI without anticoagulation appears to be a safe and effective strategy for performing PCI. This antiplatelet-focused approach-predicated upon platelet slip-deserves further investigation. If validated, it should result in a paradigm shift in antithrombotic strategies used during PCI and an evidence-based change in the PCI guidelines.
SJD: Both conception and design as well as acquisition, analysis and interpretation of data; both drafting of the manuscript and revising it critically for important intellectual content; final approval of the manuscript submitted; and agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
AP: Analysis and interpretation of data; revising manuscript critically for important intellectual content; final approval of the manuscript submitted; and agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
CLG: Analysis and interpretation of data; revising manuscript critically for important intellectual content; final approval of the manuscript submitted; and agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
LOP: Analysis and interpretation of data; revising manuscript critically for important intellectual content; final approval of the manuscript submitted; and agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
JET: Both conception and design as well as analysis and interpretation of data; revising manuscript critically for important intellectual content; final approval of the manuscript submitted; and agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
None.
None.
The authors declare no competing interests.
- Virani SS (2021) Heart disease and stroke statistics-2021 update: A report from the American Heart Association. Circulation 143: e254-e743. [Crossref]
- O’Gara PT (2013) ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127: e362-425. [Crossref]
- Anderson JL (2013) 2012 ACCF/AHA focused update incorporated into the ACCF/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127: e663-828.
- Grüntzig A (1978) Percutaneous transluminal dilatation of chronic coronary stenoses. First experiences. Schweiz Med Wochenschr 108: 1721-1723. [Crossref]
- Sherwood MW, Tcheng JE (2015) Eptifibatide in coronary intervention: past time for the next chapter. Circ Cardiovasc Interv 8: e002340.
- Tricoci P (2013) Cardiac troponin after percutaneous coronary intervention and 1-year mortality in non-ST-segment elevation acute coronary syndrome using systematic evaluation of biomarker trends. J Am Coll Cardiol 62: 242-251. [Crossref]
- Park DW (2013) Frequency, causes, predictors, and clinical significance of peri-procedural myocardial infarction following percutaneous coronary intervention. Eur Heart J 34: 1662-1669. [Crossref]
- Mehran R (2009) Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 30: 1457-1466.
- Mehran R (2011) Standardized bleeding definitions for cardiovascular clinical trials: a consensus report from the Bleeding Academic Research Consortium. Circulation 123: 2736-2747. [Crossref]
- Topol EJ (1993) Use of a direct antithrombin, hirulog, in place of heparin during coronary angioplasty. Circulation 87: 1622-1629.
- Xu XR (2016) Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci 53: 409-430.
- Sang Y, Roest M, de Laat B, de Groot PG, Huskens D (2020) Interplay between platelets and coagulation. Blood Rev 12: 100733.
- Vogler EA, Siedlecki CA (2009) Contact activation of blood-plasma coagulation. Biomaterials 30: 1857-1869.
- Aldenhoff YB, Hanssen JH, Knetsch ML, Koole LH (2007) Thrombus formation at the surface of guide-wire models: effects of heparin-releasing or heparin-exposing surface coatings. J Vasc Interv Radiol 18: 419-425.
- Yau JW (2011) Mechanism of catheter thrombosis: comparison of the antithrombotic activities of fondaparinux, enoxaparin, and heparin in-vitro and in-vivo. Blood 118: 6667-6674. [Crossref]
- Smith SC (2006) ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/SCAI Writing Committee to Update the 2001 Guidelines for Percutaneous Coronary Intervention). American Heart Association Web Site. Available at: http://www.americanheart.org. Circulation 113: 156-175.
- Denardo SJ (2020) Validated model of platelet slip at stenosis and device surfaces. Platelets 31: 373-382.
- Denardo SJ, Davis KE, Tcheng JE (2005) Elective percutaneous coronary intervention using broad-spectrum antiplatelet therapy (eptifibatide, clopidogrel, and aspirin) alone, without scheduled unfractionated heparin or other anti-thrombin therapy. Am Heart J 149: 138-144. [Crossref]
- Valencia R (2007) Efficacy and safety of triple antiplatelet therapy with and without concomitant anticoagulation during elective percutaneous coronary intervention (the REMOVE trial). Am J Cardiol 100: 1099-1102.
- Stabile E (2008) The CIAO (Coronary Interventions Antiplatelet-based Only) Study: a randomized study comparing standard anticoagulation regimen to absence of anticoagulation for elective percutaneous coronary intervention. J Am Coll Cardiol 52: 1293-1298. [Crossref]
- Denardo SJ, Davis KE, Tcheng JE (2007) Effectiveness and safety of reduced-dose enoxaparin in non-ST-segment elevation acute coronary syndrome followed by antiplatelet therapy alone for percutaneous coronary intervention. Am J Cardiol 100: 1376-1382.
- Acharya T (2017) In-Hospital outcomes of percutaneous coronary intervention in America's safety net: Insights from the NCDR Cath-PCI Registry. JACC Cardiovasc Interv 10: 1475-1485.
- Shimony A, Zahger D, Ilia R, Shalev A, Cafri C, et al. (2010) Impact of the community's socioeconomic status on characteristics and outcomes of patients undergoing percutaneous coronary intervention. Int J Cardiol 144: 379-382. [Crossref]
- https://www.census.gov/quickfacts/fact/table/robesoncountynorthcarolina,NC/PST045216. Accessed March 30, 2017.
- US Census Bureau, Small Area Income and Poverty Estimates, 2014. Accessed March 30, 2017.
- http://www.schs.state.nc.us/data/vital/lcd/x/heartdisease.html. x=2011, 2012, 2013, 2014, 2015, 2016, 2017. Accessed April 12, 2021.
- University of Wisconsin Population Health Institute. County Health Rankings & Roadmaps 2011-2017. www.countyhealthrankings.org. Accessed January 1, 2019.
- Denardo SJ. 2009. Exclusive antiplatelet therapy for percutaneous coronary intervention. J Am Coll Cardiol 53: 1921-1922.
- Denardo SJ, Davis KE, Reid PR, Tcheng JE (2001) Minimal dose heparin with abciximab in coronary intervention: efficacy and safety of a novel heparin dosing strategy. In: Lewis BS, editor. Advances in Coronary Artery Disease: Proceedings of the 4th International Congress on Coronary Artery Disease: Prague, Czech Republic, October 21-24, 2001. Bologna, Italy: Monduzzi Editore, 573-578.
- Bazzi S, Fiszbein M, Gebresilasse M (2020) Frontier culture: The roots and persistence of “rugged individualism” in the United States. Econometrica 88: 2329-2368.
- Smith SC (2001) ACC/AHA guidelines for percutaneous coronary intervention (revision of the 1993 PTCA guidelines)-executive summary: a report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee to Revise the 1993 Guidelines for Percutaneous Transluminal Coronary Angioplasty) endorsed by the Society for Cardiac Angiography and Interventions. Circulation 103: 3019-3041. [Crossref]
- Protection of human subjects; Belmont Report: Notice of report for public comment. 1979. Fed Regist 44: 23191-23197.
- Ryan TJ (1993) Guidelines for percutaneous transluminal coronary angioplasty. A report of the American Heart Association/American College of Cardiology Task Force on Assessment of Diagnostic and Therapeutic Cardiovascular Procedures (Committee on Percutaneous Transluminal Coronary Angioplasty). Circulation 88: 2987-3007.
- Thygesen K (2018) Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). Circulation 138: e618-e651.
- Harrington RA (2009) Platelet inhibition with cangrelor in patients undergoing PCI. N Engl J Med 361: 2318-2329.
- Montalescot G (2009) Immediate vs delayed intervention for acute coronary syndromes: a randomized clinical trial. JAMA 302: 947-954.
- Stone GW (2007) Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 369: 907-919.
- McEntegart MB (2012) Intraprocedural thrombotic events during percutaneous coronary intervention in patients with non-ST-segment elevation acute coronary syndromes are associated with adverse outcomes: analysis from the ACUITY (Acute Catheterization and Urgent Intervention Triage Strategy) trial. J Am Coll Cardiol 59: 1745-1751. [Crossref]
- Ndrepepa G (2018) Comparative prognostic value of postprocedural creatine kinase myocardial band and high-sensitivity troponin T in patients with non-ST-segment elevation myocardial infarction undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 91: 215-223.
- Pride YB (2012) Association between angiographic complications and clinical outcomes among patients with acute coronary syndrome undergoing percutaneous coronary intervention: an EARLY ACS (Early Glycoprotein IIb/IIIa Inhibition in Non-ST-Segment Elevation Acute Coronary Syndrome) angiographic substudy. JACC Cardiovasc Interv 5: 927-935.
- Stone GW (2006) Bivalirudin for patients with acute coronary syndromes. N Engl J Med 355: 2203-2216.
- Ritsinger V, Saleh N, Lagerqvist B, Norhammar A (2015) High event rate after a first percutaneous coronary intervention in patients with diabetes mellitus: results from the Swedish coronary angiography and angioplasty registry. Circ Cardiovasc Interv 8: e002328.
- Spiel AO, Gilbert JC, Jilma B (2008) Von Willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation 117: 1449-1459. [Crossref]
- Jennings LK (2009) Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost 102: 248-257.
- Hanson SR, Tucker EI (2013) Blood Coagulation and Blood-Materials Interactions, Editor(s): Buddy D. Ratner, Allan S. Hoffman, Frederick J. Schoen, Jack E. Lemons, Biomaterials Science (Third Edition), Academic Press, 2013, Pages 551-557, Chapter II.2.6 ISBN 9780123746269, https://doi.org/10.1016/B978-0-08-087780-8.00048-6. (http://www.sciencedirect.com/science/article/pii/B9780080877808000486).
- Leunissen TC (2017) The effect of P2Y12 inhibition on platelet activation assessed with aggregation- and flow cytometry-based assays. Platelets 28: 567-575.
- Valgimigli M (2014) Usefulness and safety of vorapaxar in patients with non-ST-segment elevation acute coronary syndrome undergoing percutaneous coronary intervention (from the TRACER Trial). Am J Cardiol 114: 665-673.
- Cadroy Y, Horbett TA, Hanson S (1989) Discrimination between platelet-mediated and coagulation mediated mechanisms in a model of complex thrombus formation in vivo. J Lab Clin Med 113: 436-448.
- Kajiya F (1993) Velocity profiles and phasic flow patterns in the non-stenotic human left anterior descending coronary artery during cardiac surgery. Cardiovasc Res 27: 845-850. [Crossref]
- Strony J, Beaudoin A, Brands D, Adelman B (1993) Analysis of shear stress and hemodynamic factors in a model of coronary artery stenosis and thrombosis. Am J Physiol 265: H1787-1796.
- Krams R (1999) Effect of catheter placement on 3-D velocity profiles in curved tubes resembling the human coronary system. Ultrasound Med Biol 25: 803-810.
- Back LH (1994) Estimated mean flow resistance increase during coronary artery catheterization. J Biomech 27: 169-175.
- Friedman LI, Liem H, Grabowski EF, Leonard EF, McCord CW, et al. (1970) Inconsequentiality of surface properties for initial platelet adhesion. Trans Am Soc Artif Intern Organs 16: 63-73.
- Slepian MJ (2017) Shear-mediated platelet activation in the free flow: Perspectives on the emerging spectrum of cell mechanobiological mechanisms mediating cardiovascular implant thrombosis. J Biomech 50: 20-25. [Crossref]
- Chen JY (2019) Association of parenteral anticoagulation therapy with outcomes in Chinese patients undergoing percutaneous coronary intervention for non-ST-segment elevation acute coronary syndrome. JAMA Intern Med 179: 186-194.
- Bonello L (2015) Comparison of ticagrelor versus prasugrel to prevent periprocedural myonecrosis in acute coronary syndromes. Am J Cardiol 116: 339-343.
- Gao C (2011) Heparin promotes platelet responsiveness by potentiating αIIbβ3-mediated outside-in signaling. Blood 117: 4946-4952.
- Xiao Z, Theroux P (1998) Platelet activation with unfractionated heparin at therapeutic concentrations and comparisons with a low-molecular weight heparin and with a direct thrombin inhibitor. Circulation 97: 251-256.
- Ashby DT (2003) Relation between the degree of procedural anticoagulation and complications after coronary stent implantation. Am J Cardiol 92: 319-322. [Crossref]
- Smith AJ (1996) Transient thrombotic state after abrupt discontinuation of heparin in percutaneous coronary angioplasty. Am Heart J 131: 434-439.
- Montalescot G (2014) Effect of prasugrel pre-treatment strategy in patients undergoing percutaneous coronary intervention for NSTEMI: the ACCOAST-PCI study. J Am Coll Cardiol 64: 2563-2571.
- Mathews R (2011) In-hospital major bleeding during ST-elevation and non-ST-elevation myocardial infarction care: derivation and validation of a model from the ACTION Registry®-GWTG™. Am J Cardiol 107: 1136-1143.
- Tousek P (2017) Incidence, treatment strategies and outcomes of acute coronary syndrome with and without ongoing myocardial ischaemia: results from the CZECH-3 registry. Eur Heart J Acute Cardiovasc Care 8: 687-694.
- Alexander KP (2005) Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 294: 3108-3116. [Crossref]
- Tolleson TR (2003) Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 41: 386-393.
- Lauer MA (2001) Attenuation of rebound ischemia after discontinuation of heparin therapy by glycoprotein IIb/IIIa inhibition with eptifibatide in patients with acute coronary syndromes: observations from the platelet IIb/IIIa in unstable angina: receptor suppression using integrilin therapy (PURSUIT) trial. Circulation 104: 2772-2777. [Crossref]
- Ibrahim K (2018) Fentanyl delays the platelet inhibition effects of oral ticagrelor: Full Report of the PACIFY Randomized Clinical Trial. Thromb Haemost 118: 1409-1418.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/017029s108lbl.pdf. Accessed January 1, 2019.
- Seto AH (2018) Length of stay following percutaneous coronary intervention: An expert consensus document update from the society for cardiovascular angiography and interventions. Catheter Cardiovasc Interv 92: 717-731.
- Sandoval Y, Bell MR, Gulati R (2019) Transradial artery access complications. Circ Cardiovasc Interv 12: e007386.
- Gaudino M (2020) Association of radial artery graft vs saphenous vein graft with long-term cardiovascular outcomes among patients undergoing coronary artery bypass grafting: A systematic review and meta-analysis. JAMA 324: 179-187. [Crossref]
- Tamez H (2018) Cost implications of intraprocedural thrombotic events and bleeding in percutaneous coronary intervention: Results from the CHAMPION PHOENIX ECONOMICS Study. Catheter Cardiovasc Interv 92: E348-E355.
- Hershey D, Cho SJ (1966) Blood flow in rigid tubes: thickness and slip velocity of plasma film at the wall. J Appl Physiol 21: 27-32.
- Kumar A (2018) High coronary shear stress in patients with coronary artery disease predicts myocardial infarction. J Am Coll Cardiol 72: 1926-1935. [Crossref]
Editorial Information
Editor-in-Chief
Terry Lichtor
Tsuyoshi Hirata
Shinya Mizuno
Giacomo Corrado
Article Type
Review Article
Publication history
Received: March 26, 2021
Accepted: April 21, 2021
Published: April 23, 2021
Copyright
©2021 Denardo SJ. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
Scott J. Denardo, Alice Parish, Cynthia L. Green, Latasha Oxendine-Phillips and James E. Tcheng, et al. (2021) Antiplatelet therapy only, without anticoagulation, for percutaneous coronary intervention: A translational application of platelet slip 7: DOI: 10.15761/JTS.1000455.
Figure 1. Slip of microspheres in a cylindrical scaled-up model of coronary flow (longitudinal view of horizontal middle plane of hemi-cylinder) [17].
The velocities of 3.12 µm diameter polystyrene microspheres-modelling platelets and as determined by laser-Doppler velocimetry (blue rings and discs: ○;●, respectively)-are compared with COMSOL computer-simulated plasma velocity profiles (black solid lines). The model contains a central solid cylinder representing a PCI guide wire (a “cylinder-within-a-cylinder”), a 20% stenosis by CSA, and the RE of 300 is typical for coronary flow [49]. All velocities are normalized to the mean velocity u0, calculated from direct volumetric measurement of bulk fluid flow. Computer simulations apply the no-slip boundary condition at each surface, as the wall shear stresses in the unobstructed coronary circulation promote minimal plasma slip [73,74]. The blue discs (●) show effective slip (i.e.: non-zero velocity) of microspheres immediately adjacent to the central cylinder surface. (CSA - cross sectional area; PCI - percutaneous coronary intervention; RE - Reynolds number)
Figure 2. Flow diagram for the report.
Of the 922 patients undergoing PCI during the report period, 481 patients were eligible for this report (with 441 patients excluded). There were 4 unsuccessful PCIs among the excluded patients: 3 involved chronic total occlusions; the other involved a target lesion within a large side branch of a major coronary artery, with the side branch behind 2 layers of stent previously deployed within the parent coronary artery (“double stent jail”). None of the 4 unsuccessful PCI patients experienced a 72-hour or 30-day adverse event. (CHF - congestive heart failure; DAPT – oral dual antiplatelet therapy; NSTEMI - non-ST segment elevation myocardial infarction; PCI - percutaneous coronary intervention; UFH - unfractionated heparin; USA - unstable angina)
Figure 3. Activity of the coagulation cascade and platelets at a PCI device surface.
The contact coagulation system (light blue shade) is initiated by contact and adsorption of factor XII (Hageman) onto device surfaces with a consequent change in its conformation producing activation (factor XIIa) [45]. Other proteins, including factor I (fibrinogen), are adsorbed by device surfaces as well. Moreover, further down the coagulation cascade, the effect of factor IIa (thrombin) on platelet activation during PCI performed in the context of NSTE-ACS is diminished by DAPT [46,47].
Panel A: In a low-shear environment, naïve platelets can adhere via the inactivated GP IIb/IIIa receptor to adsorbed factor I (and other proteins) and then become activated, secrete and aggregate. This mechanism is amplified for activated platelets. However, in a low-shear environment, the contact coagulation system dominates [48]. Platelet aggregates, in conjunction with the end-product of the coagulation cascade, factor Ia (fibrin), form thrombus.
DAPT=dual oral antiplatelet therapy; F=Coagulation factor; GP=Glycoprotein; NSTE-ACS=Non-ST segment elevation acute coronary syndrome; PCI=Percutaneous coronary intervention; vWfa=activated von Willebrand factor.
Figure 3. Activity of the coagulation cascade and platelets at a PCI device surface.
Panel B: In a high-shear environment, platelet slip at the device surface interrupts physical contact between the platelet and that surface, therefore diminishing adhesion and functionally extinguishing effects of the contact coagulation system. Thus, no thrombus forms by this mechanism. However, high shear can nonetheless activate platelets via vWfa [11,12,43,44], with ultimate thrombus formation.
DAPT=dual oral antiplatelet therapy; F=Coagulation factor; GP=Glycoprotein; NSTE-ACS=Non-ST segment elevation acute coronary syndrome; PCI=Percutaneous coronary intervention; vWfa=activated von Willebrand factor.
Table 1. Baseline characteristics of 481 report patients based upon GPI infusion duration, both before and after matching*
Baseline Characteristics |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value
(Unmatched Groups)
[SMD] |
P-value
(Matched Groups)
[SMD] |
GPI Duration:
(Sample Size) |
Age (years) |
61.9±11.7 |
62.9±11.4 |
62.4±12.6 |
0.61 [0.04] |
0.78 [0.04] |
Male sex |
226 (59.3%) |
59 (59.0%) |
54 (54.0%) |
0.34 [0.11] |
0.56 [0.10] |
Race |
|
|
|
0.30 [0.12] |
0.80 [0.05] |
Caucasian |
181 (47.5%) |
52 (52.0%) |
50 (50.0%) |
|
|
African-American |
69 (18.1%) |
25 (25.0%) |
23 (23.0%) |
|
|
Native-American |
131 (34.4%) |
23 (23.0%) |
27 (27.0%) |
|
|
Married† |
214 (56.2%) |
57 (57.0%) |
49 (49.0%) |
0.20 [0.15] |
0.30 [0.16] |
Insurance type† |
|
|
|
0.56 [0.30] |
0.60 [0.35] |
Non-private‡ |
258 (67.7%) |
69 (69.0%) |
62 (62.0%) |
|
|
Private |
80 (21.0%) |
21 (21.0%) |
25 (25.0%) |
|
|
Uninsured |
43 (11.3%) |
10 (10.0%) |
13 (13.0%) |
|
|
BMI (kg/m2) |
31.0±7.8 |
30.0±6.8) |
30.1±7.3 |
0.34 [0.12] |
0.84 [0.014] |
Major CHD risk factors: |
|
|
|
|
|
Diabetes mellitus |
181 (47.5%) |
42 (42.0%) |
51 (51.0%) |
-- |
-- |
Insulin-dependent |
89 (23.4%) |
19 (19.0%) |
23 (23.0%) |
0.72 [0.10] |
0.45 [0.13] |
Non-insulin-dependent |
92 (24.1%) |
23 (23.0%) |
28 (28.0%) |
Hypertension |
346 (90.8%) |
86 (86.0%) |
83 (83.0%) |
0.03 [0.23] |
0.71 [0.08] |
Dyslipidemia |
365 (95.8%) |
92 (92.0%) |
93 (93.0%) |
0.29 [0.12] |
1.00 [0.04] |
Cigarette smoking-active |
137 (36.0%) |
34 (34.0%) |
33 (33.0%) |
0.67 [0.06] |
0.99 [0.04] |
Family history of premature CHD† |
346 (91.1%) |
91 (91.0%) |
91 (91.0%) |
0.99 [<0.01] |
1.00 [<0.01] |
Other co-morbidities |
|
|
|
|
|
Prior MI |
161 (42.3%) |
33 (33.0%) |
35 (35.0%) |
0.19 [0.15] |
0.88 [0.04] |
Prior revascularization§ |
223 (58.5%) |
50 (50.0%) |
52 (52.0%) |
0.24 [0.13] |
0.88 [0.04] |
Cerebral vascular disease |
53 (13.9%) |
22 (22.0%) |
21 (21.0%) |
0.08 [0.19] |
1.00 [0.02] |
Peripheral vascular disease |
42 (11.0%) |
9 (9.0%) |
10 (10.0%) |
0.77 [0.03] |
1.00 [0.03] |
Chronic obstructive pulmonary disease |
141 (37.0%) |
30 (30.0%) |
31 (31.0%) |
0.27 [0.13] |
1.00 [0.02] |
Chronic renal insufficiency |
93 (24.4%) |
15 (15.0%) |
13 (13.0%) |
0.01 [0.30] |
0.83 [0.06] |
Clinical presentation: |
|
|
|
0.26 [0.12] |
0.97 [0.04] |
NSTEMI |
177 (46.5%) |
50 (50.0%) |
51 (51.0%) |
|
|
USA |
155 (40.7%) |
42 (42.0%) |
42 (42.0%) |
|
|
CHF |
49 (12.9%) |
8 (8.0%) |
7 (7.0%) |
|
|
Heart failure class at admission (NYHA) |
|
|
|
<0.01 [0.84] |
0.84 [0.19] |
1 or 2 |
133 (34.9%) |
24 (24.0%) |
21 (21.0%) |
|
|
3 |
162 (42.5%) |
58 (58.0%) |
62 (62.0%) |
|
|
4 |
86 (22.6%) |
18 (18.0%) |
17 (17.0%) |
|
|
Left ventricular ejection fraction (%) |
43.0±9.0 |
43.8±8.1 |
43.7±7.1 |
0.91 [0.09] |
0.96 [0.01] |
BMI=Body mass index, CHD=Coronary heart disease, CHF=Congestive Heart Failure (acute, and requiring intravenous diuretic therapy),
GPI=Glycoprotein inhibitor, NSTEMI=Non-ST Segment Elevation Myocardial Infarction, NYHA=New York Heart Association,
SMD=Standardized Mean Difference, USA=Unstable Angina.
*Data are presented as mean ± standard deviation or as number (percentage).
†Variable not used in propensity score matching.
‡Non-private insurance: Medicare, Medicaid or Veterans Affairs.
§Prior revascularization: Percutaneous or surgical.
P-value (Unmatched Groups): Comparison between GPI ≤4.0 hours (N=100) vs.GPI >4.0 hours-unmatched group (N=381).
P-value (Matched Groups): Comparison between GPI ≤4.0 hours (N=100) vs.GPI >4.0 hours matched group (N=100).
Table 2. PCI details based upon GPI infusion duration, both before and after matching*
PCI Details |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value
(Unmatched Groups)
[SMD] |
P-value
(Matched Groups)
[SMD] |
GPI Duration:
(Sample Size) |
GPI duration (hour)† |
11.2±4.0 |
11.2±6.8 |
3.6±0.6 |
<0.01 [2.66] |
<0.01 [1.57] |
Activated clotting time (sec.) |
145.4±16.7 |
144.7±16.7 |
142.4±18.2 |
0.21 [0.17] |
0.39 [0.13] |
Pre-PCI anticoagulation |
|
|
|
|
|
Unfractionated heparin† |
137 (36.0%) |
33 (33.0%) |
38 (38.0%) |
0.71 [0.04] |
0.52 [0.11] |
Enoxaparin |
164 (43.0%) |
48 (48.0%) |
43 (43.0%) |
0.96 [0.01] |
0.55 [0.10] |
Final Dose (mg/kg) |
0.75±0.27 |
0.74±0.27 |
0.64±0.27 |
0.02 [0.41] |
0.15 [0.37] |
Time of final dose pre-PCI (hour)† |
18.1±8.7 |
18.1±7.7 |
17.9±8.2 |
0.97 [0.02] |
0.38 [0.03] |
No anticoagulation† |
80 (21.0%) |
19 (19.0%) |
19 (19.0%) |
0.66 [0.05] |
1.00 [<0.01] |
Pre-PCI P2Y12 inhibitor‡ |
|
|
|
|
|
Already receiving pre-PCI |
263 (69.0%) |
51 (51.0%) |
53 (53.0%) |
0.03 [0.33] |
0.88 [0.04] |
Loaded at time of PCI |
164 (43.0%) |
79 (79.0%) |
79 (79.0%) |
<0.01 [0.79] |
1.00 [<0.01] |
Lesion characteristics: |
|
|
|
|
|
Multi-vessel |
105 (27.6%) |
21 (21.0%) |
23 (23.0%) |
0.36 [0.11] |
0.87 [0.05] |
De novo |
330 (86.6%) |
92 (92.0%) |
88 (88.0%) |
0.72 [0.04] |
0.48 [0.13] |
Lesion type B2 or C |
206 (54.1%) |
60 (60.0%) |
60 (60.0%) |
0.54 [0.26] |
0.95 [0.07] |
Stenosis severity
(% by diameter): |
|
|
|
|
|
Pre-PCI |
90.5±9.2 |
90.1±9.9 |
90.5±9.6 |
0.83 [<0.01] |
0.80 [0.04] |
Post-PCI |
-2.3±6.3 |
-1.3±5.9 |
-1.2±6.3 |
0.07 [0.18] |
0.95 [0.02] |
TIMI flow: |
|
|
|
|
|
Pre-PCI |
2.45±0.86 |
2.35±0.95 |
2.35±0.88 |
0.18 [0.12] |
1.00 [<0.01] |
Post-PCI |
2.96±0.20 |
2.94±0.24 |
2.95±0.22 |
0.73 [0.05] |
0.76 [0.04] |
Stent design: |
|
|
|
|
|
Drug-eluting |
213 (55.9%) |
41 (41.0%) |
43 (43.0%) |
0.02 [0.26] |
0.89 [0.04] |
Diameter (mm) |
2.56±0.45 |
2.53±0.45 |
2.53±0.51 |
0.23 [0.06] |
0.94 [<0.01] |
Length (mm) |
21.3±11.4 |
19.5±8.1 |
19.4±7.9 |
0.46 [0.19] |
0.89 [0.02] |
Maximum deployment pressure (atm) |
17.2±2.7 |
16.9±2.7 |
16.8±3.3 |
0.10 [0.13] |
0.86 [0.03] |
Procedure duration (min.)§ |
29.6±15.2 |
26.1±12.0 |
28.0±19.5 |
0.15 [0.09] |
0.44 [0.12] |
Length of stay post-PCI (days)†
Length of stay range (days)† |
1.5±1.4
1-14 |
1.5±1.7
1-14 |
1.7±3.0
1-27 |
0.48 [0.09] |
0.76 [0.08] |
GPI=Glycoprotein inhibitor, PCI=Percutaneous coronary intervention.
*Data are presented as mean ± standard deviation or as number (percentage).
†Variable not used for propensity score matching.
‡Pre-PCI P2Y12 inhibitor: If unclear whether patient fully loaded pre-PCI, then loading dose administered at that time.
§Procedure duration: Defined from the time of insertion of 1st guiding catheter (if multiple) to acquisition of final angiographic images.
P-value (Unmatched Groups): Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-unmatched group (N=381).
P-value (Matched Groups): Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-matched group (N=100).
Table 3. 72-hour outcomes based upon GPI infusion duration, both before and after matching*
Outcomes |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value |
OR (CI) |
GPI Duration:
(Sample Size) |
MACE |
11 (3.0%) |
5 (5.2%) |
1 (1.1%) |
0.22 |
0.20 (0.00-1.79) |
Death |
0 |
0 |
0 |
-- |
-- |
MI† |
11 (3.0%) |
5 (5.2%) |
1 (1.1%) |
0.22 |
0.20 (0.00-1.79) |
Q-wave |
0 |
0 |
0 |
-- |
-- |
Type 4a MI |
11 (3.0%) |
5 (5.2%) |
1 (1.1%) |
0.22 |
0.20 (0.00-1.79) |
Type 4b MI |
0 |
0 |
0 |
-- |
-- |
TVR |
0 |
0 |
0 |
-- |
-- |
TLR |
0 |
0 |
0 |
-- |
-- |
IPTE |
46 (12.1%) |
13 (13.0%) |
8 (8.0%) |
0.38 |
1.63 (0.62-4.52) |
Subsequent MACE |
9 (19.6%) |
4 (30.8%) |
1 (12.5%) |
-- |
-- |
Bleeding events |
|
|
|
|
|
BARC 2 |
4 (1.0%) |
1 (1.0%) |
0 |
|
1.00 (0.00-19.00) |
BARC 3‡ |
5 (1.3%) |
3 (3.0%) |
1 (1.0%) |
|
0.33 (0.01-4.15) |
BARC 5 |
0 |
0 |
0 |
-- |
-- |
Thrombocytopenia |
6 (1.6%) |
2 (2.0%) |
0 |
0.25 |
0.41 (0.00-3.47) |
Profound (<20,000/μl) |
0 |
0 |
0 |
-- |
-- |
BARC=Bleeding Academic Research Consortium9, BARC 2=Any overt, actionable sign of bleeding that does not fit the criteria for BARC type 3-5, BARC 3=Overt bleeding plus hemoglobin drop of 3 to <5 g/dL (provided hemoglobin drop is related to bleed); any transfusion with overt bleeding, BARC 5= Fatal bleeding.
GPI=Glycoprotein inhibitor, IPTE=Intraprocedural thrombotic events, MACE=Major adverse cardiac events (death from any cause; Type 4 myocardial infarction; target vessel revascularization), MI=Myocardial infarction, TLR=Target lesion revascularization (including both urgent and non-urgent), TVR=Target vessel revascularization.
*Data are presented as number (percentage).
†N=21 patients excluded from 72-hour analysis due to pre-PCI increasing troponin I (Unmatched N excluded=16/381; Matched GPI >4.0 hour N excluded=4/100; GPI ≤4.0 hour N excluded=5/100).
‡BARC 3 bleeding events: All type 3a.
P-value: Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-matched group (N=100).
Outcomes not mutually exclusive.
Table 4. 30-Day outcomes based upon GPI infusion duration, both before and after matching*
Outcomes |
GPI >4.0 hour
Unmatched
(N=381) |
GPI >4.0 hour
Matched
(N=100) |
GPI ≤4.0 hour
(N=100) |
P-value |
OR (CI) |
GPI Duration:
(Sample Size) |
MACE |
18 (4.7%) |
5 (5.0%) |
3 (3.0%) |
0.73 |
0.60 (0.09-3.08) |
Death |
3 (0.8%) |
0 |
1 (1.0%)† |
0.50 |
1.00 (0.05-ꝏ) |
MI‡ |
|
|
|
|
|
Type 1§+4a |
14 (3.7%) |
5 (5.0%) |
2 (2.0%) |
0.45 |
0.40 (0.04-2.44) |
Type 4b |
2 (0.5%)¶ |
0 |
0 |
-- |
-- |
TVR |
3 (0.8%) |
0 |
1 (1.0%) |
0.50 |
1.00 (0.05-ꝏ) |
TLR |
2 (0.5%) |
0 |
0 |
-- |
-- |
Bleeding events |
|
|
|
|
(compared to 0) |
BARC 2 |
6 (1.6%) |
1 (1.0%) |
0 |
0.22 |
1.00 (0.00-19.00) |
BARC 3 |
9 (2.4%) |
5 (5.0%) |
1 (1.0%) |
0.20 (0.00-1.79) |
BARC 5 |
0 |
0 |
1 (1.0%)† |
1.00 (0.05-ꝏ) |
Readmission |
72 (18.9%) |
17 (17.0%) |
12 (12.0%) |
0.41 |
0.64 (0.25-1.60) |
Principle diagnosis:
Cardiovascular |
17 (23.6%) |
5 (29.4%) |
3 (25.0%) |
0.50 |
1.00 (0.00-19.00) |
Principle diagnosis:
Non-cardiovascular |
55 (76.4%) |
12 (70.6%) |
9 (75.0%) |
BARC=Bleeding Academic Research Consortium9, BARC 2=Any overt, actionable sign of bleeding that does not fit the criteria for BARC type 3-5,
BARC 3=Overt bleeding plus hemoglobin drop of 3 to <5 g/dL (provided hemoglobin drop is related to bleed); any transfusion with overt bleeding,
BARC 5= Fatal bleeding.
GPI=Glycoprotein inhibitor, IPTE=Intraprocedural thrombotic events, MACE=Major adverse cardiac events (death from any cause; myocardial infarction; target vessel revascularization). MI=Myocardial infarction, TLR=Target lesion revascularization (including both urgent and non-urgent), TVR=Target vessel revascularization.
*Data are presented as number (percentage).
†Death on day 27 post-PCI: Documented fatal gastrointestinal bleed (Non-IPTE patient).
‡N=21 patients excluded from 72-hour analysis due to pre-PCI increasing troponin I (Unmatched N excluded=16/381; Matched GPI >4.0 hour N excluded=4/100; GPI ≤4.0 hour N excluded=5/100).
§Each NSTEMI.
¶Both events involved same Non-IPTE patient with documented medical non-compliance.
P-value: Comparison between GPI ≤4.0 hours (N=100) vs. GPI >4.0 hours-matched group (N=100).
Outcomes not mutually exclusive.