This case report describes an unusual presentation of localised tumefactive AL amyloidosis (‘amyloidoma’) of the peripheral nervous system involving the trigeminal nerve at the level of the gasserian ganglion. The patient presented with sensory and motor symptoms affecting the face due to a bilateral trigeminal neuropathy. Amyloidosis can present with single organ involvement including isolated peripheral nervous system involvement. Peripheral nerve amyloidosis is typically associated with a monoclonal gammopathy or the rarer hereditary transthyretin due to mutations in the transthyretin gene. Interestingly, the patient had a history of Bell’s palsy and we hypothesise that a chronic infection with herpes simplex virus or other neurotropic virus may have resulted in a localised inflammatory process which was the basis for the formation of the amyloidomas.
Bilateral trigeminal nerve amyloidoma is extremely rare and has only been reported twice previously. Neuroimaging revealed lesions of bilateral trigeminal nerves at the level of the gasserian ganglion involving Meckel’s cave and extending to the cavernous sinus. Following multiple biopsies, a histopathological diagnosis of a localised amyloidoma was made. Further investigations did not reveal evidence of an underlying monoclonal gammopathy or systemic amyloidosis. Localised AL amyloidosis (amyloidoma) affecting the peripheral nervous system is an unusual but important clinical manifestation of amyloidosis.
amyloidoma, gasserian ganglion, herpes simplex virus, localised amyloidosis, meckel’s cave, trigeminal
AL: Amyloid Light Chain; CN: Cranial Nerve, HSV: Herpes simplex Virus, ATTRwt: Amyloid Transthyretin Wild-Type, FAP: Familial Amyloid Polyneuropathies.
Cases of isolated amyloidomas of the peripheral nervous system are exceedingly rare [1]. Amyloidosis is now recognised as most commonly due to deposition of wild-type transthyretin (ATTRwt amyloidosis) but can occur in a variety of other inherited and non-inherited conditions. Published cases of bilateral trigeminal amyloidoma with both intraneural and extraneural involvement at the level of the gasserian ganglion with trigeminal nerve involvement is exceptionally rare with only three reported cases with bilateral involvement [2-4]. The gasserian ganglion is the sensory ganglion of the trigeminal nerve (CN V) which occupies Meckel’s cave posteriorly and anteriorly is in the lateral wall of the cavernous sinus. Most of the published cases of amyloidomas with gasserian ganglion involvement present as unilateral trigeminal neuropathies [2,3,5]. There are only 17 published cases of amyloidoma with either unilateral trigeminal nerve or gasserian ganglion involvement [6].
Localised AL (amyloid light chain) amyloidosis of the trigeminal nerve is not a commonly considered differential of a gasserian ganglion lesion. Establishing a diagnosis of localised AL amyloidosis can be difficult. Localised AL amyloidoma is not usually associated with monoclonal gammopathy or other causes of systemic amyloidosis but these underlying conditions should be considered [7].
The patient had two non-diagnostic biopsies before the third diagnostic biopsy that was used to establish the diagnosis of an amyloidoma. Hence, this case highlights the importance of repeat biopsies in the event that a definitive diagnosis cannot be made on a biopsy when confronted with diagnostic uncertainty. Various histochemical and immunohistochemical techniques were inconclusive in determining the nature of the protein causing localised amyloidosis. However, proteomic analysis using liquid chromatography tandem mass spectrometry confirmed the amyloid deposits to be comprised of immunoglobulin light chains.
This was an atypical first presentation of localised AL amyloidosis (amyloidoma) affecting the fifth cranial nerves with no known predisposition.
A 45-year-old Caucasian male from regional Australia, presented with right-sided facial tingling associated with a dull bilateral retro-orbital type pain. Over the course of the 12 months leading to presentation, the patient’s sensory symptoms worsened and more recently, involved both sides of his face including over his chin. He also experienced paraesthesia of his tongue and lips. In addition, the patient reported an 18-month history of episodic headaches described as a ‘pressure’ sensation, several times each week which was more severe at the end of the day. The headaches were not associated with migrainous features or autonomic dysfunction and were unrelated to changes in posture. The patient had some mild difficulty with swallowing and intermittently started to dribble after eating.
The patient had no history of anosmia, but he reported some mild non-specific changes in taste. The patient was systemically well with no history of fevers or weight loss. Interestingly, the patient reported a history of left-sided lower motor neuron facial weakness which had been diagnosed as a Bell’s palsy 7 years prior to presentation. This had resulted in mild residual left-sided facial weakness. There was no history of pain associated with the onset of the presumed Bell’s palsy. No neuroimaging at the time of the diagnosis of the Bell’s palsy was available.
There was no history of any significant exposure to animals. He had no family history of amyloidosis and no known family history of any neurological disorders. Background medical history included untreated obstructive sleep apnoea, childhood osteomyelitis, bipolar affective disorder and obesity. There was no known history of malignancy. The patient initially had a non-conclusive needle biopsy of the left gasserian ganglion lesion 5 months prior, so underwent a stereotactic needle biopsy of the right gasserian ganglion which also came back non-conclusive.
The patient’s current medications included lithium 750mg nocte as well as celecoxib 200mg daily. The patient had a history of chemical exposure as he previously worked as a spray painter and crop sprayer, with a 20 pack-year history of smoking. He did not have a history of excess alcohol consumption. He was independent with all basic and instrumental activities of daily living.
On examination, there was reduced sensation over the first division of CN V bilaterally (V1) as well as the second division of CN V (V2) on the left side. The left-sided corneal reflex was reduced (efferent division of the left CN V1). The rest of the cranial nerve examination was normal with no evidence facial nerve involvement or evidence of an ophthalmoplegia. The remainder of his full neurological and physical examination was normal.
The CT and MRI of the brain and skull base identified a mass lesion involving Meckel’s cave and the gasserian ganglion on the right side. There was evidence of calcification as well as a similar but less extensive lesion involving Meckel’s cave on the left side. The radiologic opinion as to the nature of these bilateral lesions was uncertain but an inflammatory lesion or chondroid neoplasm were thought possible.
The patient had two initial non-diagnostic stereotactic needle biopsies. In view of the ongoing symptoms and lack of a definitive diagnosis, a decision was made to proceed with an open biopsy. The patient underwent a diagnostic stereotactic right temporal craniotomy and biopsy of the right Meckel’s cave lesion. At surgery, the tumour was calcified, densely adherent to the Meckel’s cave and difficult to remove. The patient made an uneventful post-operative recovery. Multiple fragments of grey soft tumour were sent for histology.
The patient completed a 3-week course of low dose fractionated stereotactic radiotherapy to bilateral gasserian ganglions to prevent symptomatic progression from further growth of the amyloidomas and also provide analgesia. He will be monitored with follow-up neuroimaging to assess whether further radiotherapy is required. There are no guidelines directing surveillance neuroimaging in patients with isolated amyloidomas of the peripheral nervous system.
A non-contrast CT brain revealed a calcific, punctate lesion involving Meckel’s cave. There was a conglomerate amorphous mass infiltrating Meckel’s cave on the right side. In addition, there was a small calcific lesion involving Meckel’s cave on the left side (Figure 1A-B).
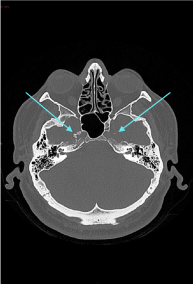
Figure 1(A): CT scan (axial section) showing a calcific, punctate lesion involving the Meckel cave on the right side. There is a small calcific lesion involving Meckel’s cave on the left side also.
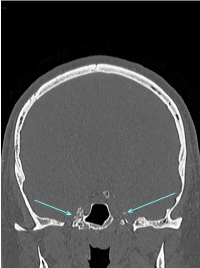
Figure 1(B): CT scan (coronal section) showing evidence of a calcific punctate lesion involving Meckel’s cave bilaterally.
The MRI brain revealed a lesion involving the Meckel’s cave on the right side measuring 11 × 20 × 10 mm. It was isointense on the T1 weighted sequence, and relatively isointense on the T2 weighted sequence (Figure 2A) with loss of the normal signal of Meckel’s cave compared to the left side. The post-contrast T1 fat saturated sequence (Figure 2B) demonstrated heterogeneous enhancement of the lesion with punctate hypointense regions within the lesion. The lesion abutted the intracavernous component of the right internal carotid artery. Most of the abnormality involved the antero-superior cavernous sinus thereby obliterating the signal from the right Meckel’s cave. There was normal enhancement of the major intracranial vascular structures. There were no other intracranial space occupying lesions and there was no mass effect. There was no meningeal enhancement that would be suggestive of leptomeningeal disease. The ventricles and CSF spaces were normal.
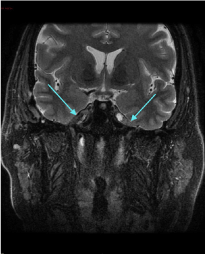
Figure 2(A): MRI scan (T2 weighted coronal sequence) showing heterogeneous enhancement of the lesion in Meckel’s cave on the right side with punctate hypointense areas within the lesion.
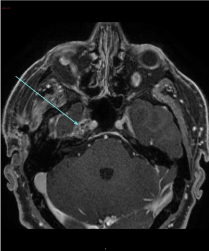
Figure 2(B): Post contrast T1 fat saturated (axial sequence) showing a lesion measuring 11 × 20 × 10 mm demonstrating heterogeneous enhancement in Meckel’s cave on the right side.
The biopsy from Meckel’s cave showed extensive amyloid deposition both within the nerve with enmeshed myelinated fibres but also in the epineural compartment (Figure 3). There were sparse plasma cells at the edge of the lesion. The extracellular proteinaceous deposit was positive for crystal violet and Congo red staining was positive with typical birefringence and dichroism effects when examined under polarised light. Potassium permanganate pre-treatment only partly obliterated staining with Congo red (Figure 4). Staining for kappa and lambda light chain was not interpretable. Immunohistochemical staining for amyloid-A was weakly positive. The histopathological diagnosis was of an amyloidoma of the trigeminal nerve at the level of the gasserian ganglion.
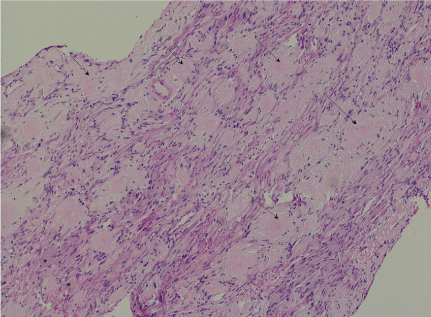
Figure 3: (H&E x100): There are amorphous pale eosinophilic deposits (arrow) amongst axons and spindled Schwann cells of the trigeminal nerve. Two ganglion cells are at the bottom left (*)
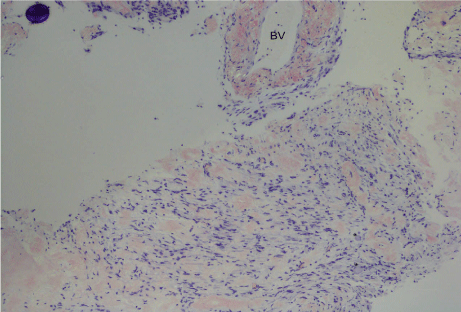
Figure 4: (Congo red x100): On polarisation, there was focal apple green birefringence of the deposits which appear red on Congo red. A blood vessel (BV) wall is involved at the top right. Two ganglion cells are at the bottom right (*).
Routine bloods tests including inflammatory markers ESR and CRP were all normal. Urine protein, creatinine and albumin/creatinine ratio were normal. Cardiac biomarkers, ECG and echocardiogram were normal, as were liver function tests. No monoclonal immunoglobin component was detected on serum and urine protein electrophoresis or the serum light free chains. A diagnostic cystoscopy was undertaken to investigate for the presence of systemic amyloidosis which was negative. As such, investigations did not reveal evidence of an underlying monoclonal gammopathy or systemic organ involvement.
Congo red deposits from the formalin fixed paraffin embedded (FFPE) bladder tissue underwent laser capture microdissection. Liquid chromatography tandem mass spectrometry was performed on peptides extracted from the dissected tissue. The results have been reanalysed with an updated database methodology. This demonstrated the amyloid forming protein immunoglobulin lambda light chain variable region as well as the amyloid associated proteins ApoE, SAP, ApoA4, vitronectin and clusterin, the latter confirming a typical amyloid proteomic signature. Small amounts of other immunoglobulin components including lambda and kappa light chain constant region, IgG, IgM and IgA constant region as well as ApoA1, gelsolin, transthyretin and fibrinogen alpha chain were also present which is a not uncommon finding of uncertain significance and may represent contaminating blood stream proteins.
The laboratory findings were consistent with a diagnosis of immunoglobulin light chain amyloidosis. Proteomic analysis cannot distinguish between localised amyloidosis and systemic amyloidosis [8], both of which are composed of immunoglobulin light chains, but in this case, there was no circulating clonal component or evidence of systemic organ involvement making the diagnosis localised AL amyloidosis.
The biopsy revealed findings consistent with an amyloidoma involving the trigeminal nerve at the level of the gasserian ganglion. It is not typically associated with systemic amyloidosis, plasma cell neoplasms, chronic inflammatory states, or chronic infections although these conditions were considered and excluded.
Although a previous biopsy performed on the contralateral side was non-diagnostic, the patient had symptomatic involvement and similar imaging abnormalities on the contralateral side and hence, in this context, it is highly likely to reflect a localised amyloidoma involving the trigeminal nerve at the level of the gasserian ganglion on the left side.
An important historical feature of the case is whether the patient’s previously diagnosed Bell’s palsy was a misdiagnosis and whether this was the first presentation of the amyloidoma causing a cranial neuropathy. This was difficult to determine as the patient did not have any dedicated neuroimaging at the time that the diagnosis of a Bell’s palsy was made. Alternatively, the Bell’s palsy may have been associated with a chronic herpes simplex virus (HSV) infection or other neurotropic virus, which resulted in a localised inflammatory process causing the formation of the amyloidomas.
Other lesions with similar neuroimaging findings include metastatic deposits and schwannomas. Some neoplasms (lymphoma and plasmacytoma) can be associated with localised amyloid formation, however there was no evidence of neoplasm in the biopsy. Potential inflammatory causes of a trigeminal neuropathy that also need to be considered as part of the differential diagnosis include Sjogren’s syndrome and vasculitis. Other connective tissue diseases that can potentially cause a progressive bilateral trigeminal neuropathy include progressive systemic sclerosis and mixed connective tissue disease.
Granulomatous conditions such as sarcoidosis also need to be considered. Inflammatory causes were investigated with appropriate laboratory investigations including ANA, ENA, ANCA and inflammatory markers. Toxins such as trichloroethylene, which is found in paints, that the patient could have been exposed to being a spray painter, can also cause trigeminal neuropathy.
The patient was diagnosed with localised AL amyloidosis of the trigeminal nerves. There was no evidence of any other systemic organ involvement and no evidence of a circulating monoclonal light chain.
A progressive bilateral trigeminal neuropathy is an unusual clinical entity and can be caused by a range of different pathological processes affecting the gasserian ganglion. The trigeminal nerve and nuclei form a complex anatomical structure in relation to the skull base and other head and neck structures. In clinical practice, it is important to distinguish trigeminal neuropathy from trigeminal neuralgia. Trigeminal neuralgia is characterised by brief attacks of pain without sensory or motor impairment. In contrast, although trigeminal neuropathy can be associated with pain, trigeminal neuropathy typically involves sensory changes, weakness and is also associated with painful mastication.
As trigeminal neuropathy worsens, it leads to loss of function in the affected nerve branches. It can be useful to think of the causes of trigeminal neuropathy as those that affect either the sensory or motor nerve branches of the trigeminal nerve, or in some instances, those causes that affect both branches of the trigeminal nerve. There are a range of disease processes which can affect the trigeminal nerves including trauma, demyelination, neoplastic, trauma, toxic and infectious causes. In some instance, the cause of trigeminal neuropathy cannot be identified but, in those cases, patients need to be monitored in view of the association of trigeminal neuropathy with malignancy and perineural infiltration. Causes of bilateral trigeminal neuralgia include:
- Infections including HSV or sinusitis
- Trauma (including iatrogenic)
- Tumours (either benign or malignant tumours which cause either compression or invasion of the trigeminal nerve)
- Other inflammatory conditions such as sarcoidosis or connective tissue disease
Trigeminal neuropathy with either unilateral or bilateral involvement can be the first presentation of malignancy. The clinical presentation and course of symptoms is generally not a useful guide to distinguish between a benign and malignant process affecting the trigeminal nerve. Even if no cause is evident at first presentation, patients need to be monitored for malignancy. Malignancy can affect the trigeminal nerve through a range of mechanisms including compression of the intra or extra-cranial part of the trigeminal nerve, perineural spread or metastasis.
One interesting speculation in this particular case was whether the diagnosis of Bell’s palsy 7 years prior to presentation was related to the bilateral trigeminal neuropathy. This is a clinical diagnosis and minimal details around how this diagnosis was initially made were available. It is believed that Bell’s palsy may be due to reactivation of HSV or other neurotropic virus, which is considered to be an acute, lower motor neuron 7th cranial nerve palsy. Additional cranial nerve involvement, including ipsilateral trigeminal nerve involvement has been seen in up to 25% of patients [9].
As far as we are aware, there have been no case reports of an association between HSV and a lower motor neuron facial palsy. It is possible that following the initial attack of Bell’s palsy, HSV lay dormant including not only the facial nucleus but also other base of skull structures including the gasserian ganglion. A viral cause of Bell’s palsy is suspected and HSV has been implicated in some case of Bell’s palsies and VZV has been implicated in some cases of Ramsay Hunt syndrome. In fact, HSV has been isolated from the gasserian ganglia in up to 50% of autopsies [10].
Following infection with a neurotropic virus, a persistent oligoclonal immunoglobulin response localised to the gasserian ganglion could have resulted in deposition of a monoclonal immunoglobulin light chain as amyloid. Thus, we hypothesise that persistent viral infection in the ganglion, that is either HSV or some other neurotropic virus caused localised AL amyloidosis. It is possible that the HSV reactivation resulted in amyloidosis in the gasserian ganglion that only presented years down the track following the diagnosis of Bell’s palsy.
This is of course is a hypothesis and it is not possible at this stage to establish causality. Although HSV has been implicated in the pathogenesis of Bell’s palsy, it has not been established with certainty and it is certainly not clear whether all lower motor neuron presentations of a facial nerve palsy are due to VZV activation. This of course raises an interesting question about a possible link between HSV or neurotropic virus latent infection and localised inflammation resulting in localised immunoglobulin synthesis and formation of an amyloidoma. That is, we hypothesise that a persistent viral infection with HSV or some other neurotropic infection was causally responsible for the localised amyloidoma.
Following the third Meckel cave biopsy, the patient’s sensory symptoms had not progressed, and he remains systemically well. The patient continues to experience intermittent bilateral facial numbness, worse on the right side associated with infrequent headaches. He has not developed any new neurological symptoms. The patient underwent a PET scan of the entire body which did not reveal any sites of increased metabolic uptake.
The mainstay of management for this condition is surgical decompression/debulking. Given the rarity of localised AL amyloidosis particularly involving cases of the peripheral nervous system, there are no clear evidence-based guidelines guiding the use of radiotherapy or chemotherapy. Amyloidomas can occasionally re-grow after resection [11]. It is unlikely that the patient will develop systemic amyloidosis [1].
Given the rarity of this disease, the natural history of condition is not clear. Further radiotherapy to the trigeminal nerve at the level of the gasserian ganglion may be required in the future should there be radiological evidence of recurrence of disease. The patient was advised to re-present to the hospital in the event of new or worsening of his neurological symptoms but to date, continues to remain clinically stable when last reviewed in the neurosurgery clinic.
Amyloid is an abnormal insoluble protein. Under certain circumstances that are currently not well understood, it can be deposited in the extracellular space. It can potentially involve almost every organ system. Deposition of amyloid in the central nervous system can be associated with a variety of conditions including neurodegenerative conditions such as cerebral amyloid angiopathy or the plaques of Alzheimer’s disease and even tumour-like deposition which is very rare. When the latter occurs, it is known as an ‘amyloidoma’. The mean age of patients with cerebral amyloidomas is 47.8 years with a slight female predominance [5].
Amyloid deposits can potentially involve any part of the peripheral nervous system including nerve trunks as well as the sensory and autonomic ganglia. It is not clear, based on studies to date, as to why amyloidosis has a predilection for certain parts of the peripheral and central nervous system. There are no clear known risk factors for localised AL amyloidosis.
Cranial neuropathies can occur as part of hereditary amyloid polyneuropathies and can be a rare but somewhat unusual complication of systemic AL amyloidosis. An amyloidoma of the trigeminal nerve is an exceptionally rare lesion. 1% of localised amyloidomas progress to develop systemic amyloidosis [1]. Most of the described cases of amyloidoma are due to AL amyloid.
Due to the absence of the blood-nerve barrier in the spinal ganglia, it is possible that intrusion of the amyloid protein into the endoneurium could occur and potentially account for a higher frequency of significant deposits of amyloid in the spinal ganglia compared to amyloid deposition in the peripheral nerves [9]. In all types of systemic amyloidosis, amyloid appears as an extracellular amorphous deposit in the epineurium, perineurium or endoneurium. Amyloid potentially causes a trigeminal neuropathy due to its involvement of the gasserian ganglion but why it has a predisposition for the gasserian ganglion is unclear.
Amyloid deposits are found mainly around blood vessels although they are closely opposed to Schwann cells or collagen fibrils in in the endoneurium. Amyloid is generally eosinophilic on H&E staining and blue on toluidine blue staining. The nerve fibres generally demonstrate axonal degeneration and the degree of fibre loss depends on the stage of the disease. During the early stages of the disease, there is a preferential loss of small myelinated and unmyelinated axons and during the later course of the disease, a diffuse pattern of axonal loss occurs. Fibre studies have demonstrated predominantly axonal degeneration with some segmental demyelination and even possibly remyelination [12].
The characteristic staining pattern of amyloid is lime-green birefringence when viewed in polarised light after staining with Congo red. This is due to the β-pleated molecular structure. There are a range of subtypes of amyloidosis. The deposits across all types consist of 7 to 10 nm unbranched rigid fibrils which are organised into an irregular array or parallel or fan-shaped fibrils. Sometimes, the fibrils can radiate stellate clusters. Amyloid components commonly contain small components of a second component corresponding to SAP which ultra-structurally, consists of thick 9 nm pentagonal discs when examined as extracts from amyloid deposits. Thus, it can be difficult to distinguish between the amyloid and other component protein constituents and at a diagnostic level, distinguishing the fibril protein composing the amyloid is possible by using immunohistochemical methods [12], or more recently with proteomic analysis.
Cranial neuropathies with amyloid involvement including trigeminal involvement can be a feature of familial amyloid polyneuropathies (FAP). Retrospective studies of patients with FAP have demonstrated significant myelinated fibre loss in the majority of affected individuals. Hence, it is important to screen affected individuals for FAP including serum and urine electrophoresis for detection of light chains. Genetic testing for FAP, especially in those individuals with a family history of neuropathy, can be considered in individuals when clinically appropriate.
When peripheral nerve involvement is suspected, a negative peripheral nerve biopsy does not necessarily exclude the possibility of amyloidosis. A whole-nerve biopsy with muscle may be required with Congo red and thioflavin staining may need to be undertaken. Depending on the level of clinical suspicion, a biopsy of another organ such as rectum, bladder or periumbilical fat may also need to be considered in the event of a negative nerve biopsy. In our patient, a bladder biopsy did not reveal any evidence of amyloidosis. Most of the studies into amyloid involve cases of peripheral nerve involvement and typically, peripheral nerve biopsies are undertaken to make a diagnosis of amyloid neuropathy. The finding of amyloid deposits in a peripheral nerve biopsy is always abnormal.
It is not clear whether the pathological changes that are seen with amyloidosis differ depending on which site of the peripheral nervous system is affected. It is also not clear whether the aetiology and pathology of amyloidosis in the case of peripheral nerve involvement is different to the pathology of amyloidosis with isolated central nervous system involvement.
In this clinical case report, we have presented a patient with a progressive bilateral trigeminal neuropathy. A biopsy of the gasserian ganglion on the right side was undertaken with histopathological findings confirming a diagnosis of a localised AL amyloidoma. Symptomatically and radiologically, the patient had bilateral symptoms consistent with a diagnosis of bilateral trigeminal nerve involvement. Hence, the most likely diagnosis was a localised bilateral AL amyloidoma of the trigeminal nerve at the level of the gasserian ganglion. Investigations for systemic amyloidosis were negative.
It is unclear whether the onset of the lower motor neuron facial weakness 7 years prior to the onset of the progressive bilateral trigeminal neuropathy was due to involvement of the facial nerve from the amyloidosis. It is most likely unrelated in view of the absence of facial nerve involvement, but this diagnostic possibility cannot be completely excluded.
Amyloidomas can affect any part of the CNS and PNS including the trigeminal nerve at the level of the gasserian ganglion. It can potentially result in an isolated cranial neuropathy or multiple cranial neuropathies. Once an amyloidoma is detected, it is important that the possibility of an associated condition as well as systemic amyloidosis is investigated and screened for, although in most cases, amyloidomas of the peripheral and central nervous system occur in isolation.
Greater awareness of this condition is needed as a potential but important differential of a gasserian ganglion lesion thereby ensuring that patients receive appropriate investigations and treatment. This clinical case highlights the importance of the appropriate use of neuroimaging including dedicated base of skull base imaging of gasserian ganglion and the cavernous sinus. An amyloidoma is an important but unusual mimic for a range of infiltrative, malignant and inflammatory lesions which can potentially affect the trigeminal nerve at the level of the gasserian ganglion and neighbouring structures. In summary, the diagnostic entity of a localised amyloidoma remains an important differential diagnosis that needs to be considered in addition to the commonly recognised infiltrative lesions and inflammatory lesions that can affect the trigeminal nerve at the level of the gasserian ganglion as well as the potential to affect other neighbouring base of skull structures.
- Localised AL amyloidosis (‘amyloidoma’) can affect a range of different sites including the peripheral nervous system in isolation without systemic involvement.
- An amyloidoma is an important but under-recognised differential diagnosis of a Meckel cave lesion apart from the more commonly recognised infiltrative and inflammatory causes.
- Amyloidosis can have either unilateral or bilateral trigeminal nerve involvement at the level of the gasserian ganglion as well as neighbouring structures including the cavernous sinus. Hence, a localised AL amyloidoma is an important differential diagnosis in a patient with a progressive trigeminal (CN V) neuropathy.
- Amyloidomas of the trigeminal nerve at the level of the gasserian ganglion are not usually associated with systemic amyloidosis.
- Amyloidomas can occur in isolation of systemic involvement which is unable to be differentiated based simply on the histopathological findings and requires further laboratory investigations and imaging.
- Treatment is targeted at local resection of the disease and radiotherapy may be required if symptoms persist and surgical resection/debulking is incomplete although the evidence base for the management and ongoing monitoring of amyloidomas is currently lacking.
The authors acknowledge the assistance of the Mater Hospital Radiology Department who kindly supplied MRI images used in this article as well as Queensland X-ray for supplying the CT images. The authors also thank Dr David Wong, Anatomical Pathologist, Mater Pathology Department for supplying the histopathology slides and for discussions around the interpretation of the histopathology.
- Mahmood S, Bridoux F, Venner CP, Sachchithanantham S, Gilbertson JA, et al. (2015) Natural history and outcomes in localised immunoglobulin light-chain amyloidosis: a long-term observational study. Lancet Haematol 21: e241-250. [Crossref]
- Kirch IE, Beaver HA, Lee AG, Green LK, Tang RA (1998) Microbial keratitis as the manifestation of trigeminal amyloidoma at initial presentation. J Neuoophthalmol 18: 192-195. [Crossref]
- O’Brien TJ, McKelvie PA, Vrodos N (1994) Bilateral trigeminal amyloidoma: an unusual case of trigeminal neuropathy with a review of the literature. J Neurosurg 81: 780-783. [Crossref]
- Gultasli N, van den Hauwe L, Bruneau M, D’Haene N, Delpierre I, et al. (2012) Bilateral Meckel’s cave amyloidoma: a case report. J Neuroradiol 39: 119-122. [Crossref]
- Gottfried ON, Chin S, Davidson HC, Couldwell WT (2007) Trigeminal amyloidoma: case report and review of the literature.Skull Base17: 317-324. [Crossref]
- Hashmi H, Dhanoa J, Manapuram S (2018) Trigeminal amyloidoma: case report and review of literature. Cureus 10: e3795. [Crossref]
- Westermark P (2012) Localized AL amyloidosis: a suicidal neoplasm?Ups J Med Sci117: 244-250. [Crossref]
- Gertz MA (2018) Immunoglobin light chain amyloidosis diagnosis and treatment algorithm. Blood Cancer J 8:44.
- Benatar M, Edlow J (2004) The spectrum of cranial neuropathy in patients with Bell's palsy.Arch Intern Med164: 2383-2385. [Crossref]
- Rapp F, Stevens JG (2019) Oncogenic Herpesviruses. Boca Raton (FL): CRC Press. Vol. 2, Chapter 1, Herpetic latency and reactivation; p. 74.
- Gandhi D, Wee R, Goyal M (2003) CT and MR imaging of intracerebral amyloidoma: case report and review of the literature.AJNR Am J Neuroradiol24: 519-522. [Crossref]
- Love S, Budka H, Ironside JW, Perry A (2015) Greenfield’s Neuropathology (9th ed). Boca Raton (FL): CRC Press.