Mucoobstructive lung diseases, including chronic obstructive pulmonary disease, asthma, cystic fibrosis, non-cystic bronchiectasis and, primary ciliary dyskinesia, are characterized by intrapulmonary accumulations of hyperconcentrated mucus. Clearance of intrapulmonary mucus by the high-velocity airflow generated by cough is the major rescue clearance mechanism in subjects with mucoobstructive diseases. Ultimately, the mucus accumulation in diseases reflects the failure of said clearance mechanism. Given the multifunctionality of mucus, the various mechanisms of mucus production and exposure to the environment, but also to the systemic circulation, it is not surprising that both its genesis and its operation can be compromised in said pathologies. We proceed to review the biochemical and biophysical properties of mucus relative to airway function, the mucin concentration in health and diseases and the integrated cilia and cough-dependent mucus clearance. Then we proceed to review various diseases that have mucus dysfunction in common, the mechanism that generates this dysfunction and how it impacts the symptoms and the natural evolution of these pathologies. At the end it is discussed how restoration of cough efficacy may be most effectively provided by restoring mucus concentrations to normal ranges with hydrating agents coupled with viscosity-lowering agents.
mucus, mucoobstructive lung diseases, mucociliary clearance, the cough
In healthy persons, a well-hydrated mucus layer is transported rapidly from the distal airways toward the trachea. In muco-obstructive diseases, epithelial defects in ion–fluid transport, mucin secretion, mucociliary clearance, or a combination of these lead to hyperconcentrated (dehydrated) mucus, failed mucus transport, and mucus adhesion to airway surfaces. Mucus that is accumulated in the trachea can be expectorated by cough as phlegm or sputum. Mucus in the small airways cannot be cleared by cough and accumulates, forming the nidus for airflow obstruction, infection, and inflammation [1]. A spectrum of lung diseases that affect the airways, including chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), primary ciliary dyskinesia, non–cystic fibrosis bronchiectasis, and bronchial asthma can be characterized as muco-obstructive diseases [2-5]. These diseases have the clinical features of cough, sputum production, and episodic exacerbations. COPD is characterized by a progressive obstruction to airflow and destruction of the lung parenchyma due to an inflammatory response and tissue damage, caused by chronic exposure to environmental factors (tobacco or biomass/fuels) in genetically susceptible individuals. Hyperproduction of mucus from the corresponding airway structures is responsible, in part, for obstruction to airflow [6]. Despite the prominence of mucus plugs in the pathophysiology of airflow obstruction in acute severe (fatal) asthma, the role of mucus plugs in the pathophysiology of airflow obstruction in chronic severe asthma is poorly understood. This limited understanding is a barrier to rational treatment of airflow obstruction in severe asthma because mucus plugs represent a tractable treatment target if they can be shown to be a cause of obstruction [7].
Non-cystic fibrosis bronchiectasis is defined clinically by the symptoms of persistent or recurrent bronchial infection related to dilated bronchi and irreversible damaged. The condition is persistent or progressive and the bronchial wall is not only long but also thickened [8]. The typical profile of bronchiectasis in adults is a long-standing productive cough (with mucus), rhinosinusitis, and fatigue in nonsmokers, with crackles upon auscultation [9]. The abnormal concentration of mucus in the lungs of patients with cystic fibrosis reflects a primary abnormality of airway epithelial ion transport [10]. In this disease, the airway epithelium is vulnerable to fluid hyperabsorption because of a defect in the secretion of chloride and bicarbonate anions mediated by mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) and by an intact sodium absorptive path. Primary ciliary dyskinesia is a genetically recessive, heterogeneous disorder of cilia motility with chronic oto-sinus-pulmonary disease and organ laterality defects in 50% of cases [11]. It is obvious that the mechanisms that lead to mucus accumulation in each of the entities are different. This work aims to give clinicians a concise and practical review of the subject.
Mucus can be defined as a biological secretion made highly viscous by the presence of gel-forming mucins. Airway mucus is approximately isotonic with serum, consisting of 90–98% water, ions (at levels similar to plasma), gel-forming mucins, and a wide array of other proteins, peptides, and small molecules [12]. Adhesion forces refers to the binding of mucus to the cell surface of the respiratory epithelium and cohesion forces to the binding of the film formed by mucus-mucus junctions, which are what keep the mucus compact. To be efficient, the high airflow of cough that clears mucus requires that you overcome both forces detaching the mucus from the surface of the airway and fracturing the mucus-mucus junctions. This facilitates mucociliary clearance. Both forces are pH-independent, therefore adding bicarbonate to the hydration mixture with saline or hypertonic sodium chloride (for example, nebulized) has little effect [13]. The mucus is really made up of two hydrogels on the airway surface (i.e., the mucus layer and the periciliary layer) that compete for hydration as a function of their relative osmotic pressures (Figure 1). A gel has both liquid and solid properties. It is a soft, elastic and deformable solid, but it also has viscous fluid characteristics. Many of the terminal sugars have carboxyl and sulfate groups and are highly anionic, allowing it to interact with matter present in the airway. Bronchial mucus normally has the consistency of egg whites. 20 genes in the genome encode mucin synthesis [14].
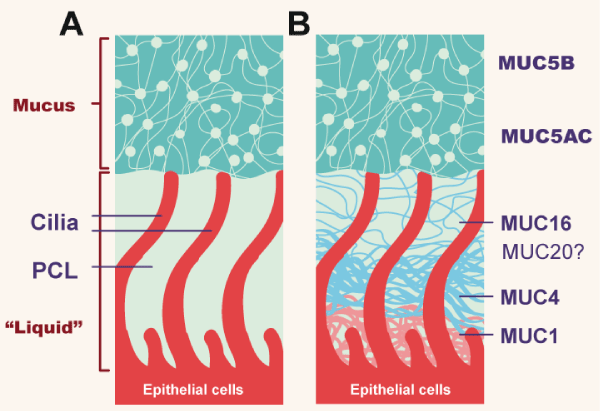
Figure 1. Topography of mucus clearance systems
(A) Classic mucus layer “floating” on periciliary liquid layer. (B) Two-gel formulation, with secreted mucins (MUC5B and MUC5AC) interpenetrating and residing over a brush-like periciliary gel layer (PCL) composed of MUC1, 4,16, and possibly 20, tethered to cilial and epithelial surfaces. (Adapted from Button B).
The mucous layer is rich in mucins, in addition to proteins, salt and water and is the one that includes pathogens and foreign material (for example, particles). Efficient clearance depends on ionic balance, water transport between mucus and epithelium, mucin secretion, and ciliary beat. There are approximately 200 cilia per cell, which beat 15-20 times per second and propel the gel at a speed of 1 mm / minute under normal conditions. If any of the components of the mucociliary apparatus is altered, the clearance deteriorates [1,14]. The two most important mucins on this sheet are MUC5B and MUC5AC. MUC5B is extensively expressed in superficial airway epithelial cells, and furthermore, in submucosal glands and occurs in both large (> 2mm) and small (<2mm) pathways. MUC5AC is produced especially on large airways. The production of both is a property of cells that express secretory proteins [15]. The most important segretagogue molecule is ATP, which acts on the apical membrane receptor P2Y2 [14]. In major airways, mucins come from submucosal glands and from goblet cells on the epithelial surface, but Clara cells are the main mucus producers in small airways. The submucosal glands are positioned to produce abundant mucus in those regions of the airways (nasal cavities, upper airways, airway bifurcations) where there is an increased probability of large particle deposition [16]. The submucosal glands are the simplest exocrine glands that exist. The mucins that form the gel are large glycosylated proteins, which form polymers with molecular weights of 2-50 MDa [14].
The periciliary layer (PCL) underlies the mucous sheet and is in contact with the epithelial surface and cilia of the airway. Between both sheets there is water. This sheet is a dense gel, formed by polymeric mucins that include MUC1, MUC4, and MUC 16 attached to the epithelial surface and cilia, in addition to other glycoproteins [12]. In healthy people it is the most concentrated hydrogel, functioning as a good moisturizer and lubricant of the airway [13], guaranteeing ciliary activity and adequate transport of the overlying mucus sheet [17]. The mucus hydration is a function of the epithelial transport of ions and water. The epithelium produces active secretion of chlorine and bicarbonate into the airway lumen (Figure 2). Both anions generate a lumen-negative voltage that draws sodium into the lumen, forming sodium chloride, and consequently the water flow towards the lumen. Calcium and ATP have a synergistic effect on this active epithelial secretion [16]. The absorption or inhibition of sodium is regulated through an epithelial sodium ion channel and the extrusion of chlorine through CFTR (cystic fibrosis transmembrane conductance regulator) and other calcium-dependent chlorine channels. Therefore, under normal conditions, the release of chlorine towards the lumen is promoted, the absorption of sodium from the lumen is inhibited and therefore the flow of water towards the lumen, hydrating the gel sheets [14]. PCL is highly permeable to water and the volume of fluid is determined by the amount of sodium chloride in the lumen of the airway.
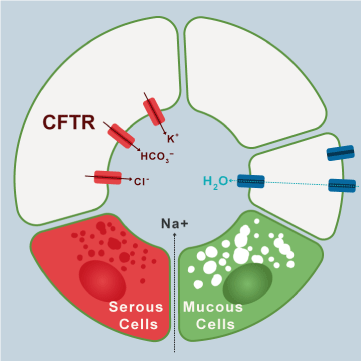
Figure 2. Ions transport mechanism in airways cells
The imagen shows clockwise the transport of electrolyte and water to the lumen of the airway in normal conditions. (Modified from Lee and Foskett).
In patients with chronic bronchitis, sustained stimulation on submucosal glands, goblet cells, and Clara cells leads to an increased amount of mucins, thickening the mucus and compromising transport [18]. High concentrations of mucins have been associated with airway disease, based exclusively on symptoms (including phlegm) without obstruction to air flow [19]. Indeed, measurements of mucins in mucus could serve as biomarkers of chronic bronchitis and be therapeutic targets in the future [13]. In CF the malformed chlorine channel reduces active secretion into the light, concentrating it on the epithelial surface, sodium is absorbed by the corresponding channel to compensate for the increased anionic charge, and water is therefore absorbed from the light, reducing hydration and concentrating mucus. The increased osmotic pressure of the mucous sheet, either due to excess mucins or because the mucus is dehydrated, compresses the PCL sheet, producing stasis and adhesion, becoming inflamed and infected [20]. When mucociliary clearance fails, cough becomes the backup mechanism to clear mucus airway. Both mechanisms depend on the properties of mucus; namely friction, viscocity, cohesion and adhesion. When both mechanisms fail, muco-obstructive diseases appear.
Important shared changes of mucoobstructive diseases are heterogeneity, onset in small pathways, frequent viral and / or bacterial infections, and cyclical exacerbations. Heterogeneity means that there are regions of the lung that are normal and other regions within the same lung that are severely diseased. A common feature that unifies muco-obstructive diseases is the early manifestation of disease in small airways (bronchioles), as evidenced by pathological examination, microcomputed tomography, and pulmonary-function studies [12]. There are two mechanisms to protect the airway against infections; mucociliary clearance and antibacterial mechanism through proteins and peptides that are secreted into the mucus by epithelial cells. Cationic antimicrobial polypeptides are responsible for bactericidal activity. Lactoferrin, lyzozyme, and secretory leukoprotease inhibitor (SLPI) are hydrophobic at physiological pH, allowing them to insert into the microbial membranes [21]. The ability of antimicrobial molecules and antibodies to suppress bacterial-infection replication is short lived, probably on the order of hours [22]. The reduced mucociiar clearance as a single factor may not be sufficient for the production of muco-obstructive diseases and requires that the mucous sheet does not move, so that mucous patches are formed, mucus plugs are formed and obstruction is generated [23].
Mucus plaques and impaction reflect an increase in mucin secretion (in turn stimulated by repetitive infections), associated with poor mucus hydration. The static and hyperconcentrated mucus becomes inflamed, generating a vicious circle of inflammation, hypersecretion, mucoid impact and new inflammation. In fact, this circle is associated with high levels of neutrophilic elastase in the airway. Neutrophilic elastase is a 29 KD serine protease that accumulates in polymorphonuclear (PMN) azurophilic granules and is released during their degranulation. In bronchiectasis, high sputum levels are associated with increased disease activity, decline in pulmonary function, and risk of exacerbations. The enzyme is proinflammatory, reduces the ciliary beat and is elevated in the sputum of what has been called "neutrophilic lung disease" (bronchiectasis, COPD, CF). In normal conditions it is inhibited by antiproteases (a-1 antitrypsin and SLPI). Its dysregulated action contributes to the pathogenesis and progression of this group of neutrophilic diseases [24,25]. Bacterial infection is a common feature of muco-obstructive lung diseases. The major site of infection in these entities is the impacted mucus plugs and the intraluminal mucus plates and not the epithelial surface of the airways. In CF, P. aeuroginosa produces alginates in the hypoxic mucus and forms macrocolonies that allow them to evade host defenses, generating chronic destructive lung [26].
These anoxia conditions favor opportunistic anaerobic infections. It is well recognized that almost all individuals microaspirate during sleep [27]. But under normal conditions these bacteria are cleared by the mucociliar system. Oral bronchoaspirated anaerobes in this impacted mucus ferment mucin producing carbon, as well as amino acids, lipids, and sugars providing nutrients for pathogens [28]. The most frequent anaerobic species are Prevotella sp, Actinomyces, Propionobactrium and Veillonella spp. Most of those anaerobes inhabit the oropharynx. In turn, infections with Pseudomona aeuroginosa rapidly reduce oxygen levels in the micro-ecosystem, favoring anaerobic infection. Chronic bacterial lung infection leads to irreversible damage to lung function and is the leading cause of death in CF, with more than 95% of deaths from respiratory failure. Using anaerobic antibiotics could improve the management of these patients [29,30]. Analyses of microbiome data from patients with cystic fibrosis or COPD have identified oral anaerobes as the first bacterial pathogens in the muco-obstructed lung [31]. This process may lead to a local immune response that contributes to the persistent inflammation seen in patients with COPD, which persists even after exposure to noxa is discontinued [32,33]. The assault produced by cigarette smoke in the intestinal microbiota can affect the host's immune system and this may have an impact on COPD. This is a fertile area of research.
Exacerbations are typical of muco-obstructive diseases. These exacerbations have a profound impact on the natural evolution of muco-obstructive pathologies, accelerate the loss of lung function, deteriorate health-related quality of life, increase the consumption of health resources and conventional hospitalizations, the use of critical care beds and increase mortality. Therefore, they are primary therapeutic targets in these diseases, and exacerbations must be treated vigorously to limit permanent loss of lung function. An exacerbation is broadly defined as a change in the patient’s perception of well-being, the seeking of health care, or a health care–implemented change in the patient’s medical regimen [34]. Common to all muco-obstructive diseases, previous rates of exacerbation and gastric aspiration are predictors of future rates of exacerbation — that is, a patient with many past exacerbations is more likely to have future exacerbations than a patient without such a history [3]. Some exacerbations are caused by an intensification of disease in areas with preexisting disease [35,36] and in another, they are associated with the spread of disease to previously unaffected regions [37]. In contrast to the sterile airways of normal lungs, bacterial pathogens are often isolated from the airways in stable COPD. This "colonization" of the tracheobronchial tree, currently believed to be innocuous, could serve as an inflammatory stimulus, independent of current tobacco smoke exposure. Bacterial colonization is associated with neutrophilic airway lumen inflammation in ex-smokers with COPD and could contribute to progression of airway disease. Gastric aspiration may be a common mechanism for spread and perhaps the major trigger for spread is viruses aspirated from upper airways into the lung, and this concept is consistent with data from patients with COPD, non–cystic fibrosis bronchiectasis, or cystic fibrosis [38-40].
Various types of stimulation, such as cigarette smoking, induce abundant production of reactive oxygen species (ROS) and proteases, which activate epidermal growth factor receptor (EGFR), Toll receptor, and other receptors, in turn triggering multiple signaling pathways such as MAPK, ERK, and NF-κβ. These pathways induce goblet cell metaplasia and hyperplasia in the airway, leading to synthesis and secretion of excessive mucin and thus to airway mucus hypersecretion [41,42]. In this way, inflammation, oxidative stress, protease imbalances, cholinergic nerve dysfunction, and other pathophysiological mechanisms can influence the risk of airway mucus hypersecretion. In addition to environmental stimulants, genetic susceptibility factors can also influence such risk [43].
Bronchial asthma
Bronchial asthma is the most common chronic inflammatory disease in children and adults therefore it contributes significantly to the casuistry of muco-obstructive diseases [44]. Bronchial asthma affects 10% of the adult population in different countries [45]. An estimated 315 million affected people worldwide and 346.000 deaths every year. Autopsy studies in Germany as early as 1880 identified mucus plugs as the cause of death in asthma and this has been a consistent finding over time [46]. However, in non-fatal asthma, there are other significant contributing factors to airflow obstruction such as plasma extravasation, smooth muscle contraction, and thickening of the airway wall, in addition to mucus hypersecretion [47]. The relationship between eosinophil granules and mucus plug formation has recently been dissected. EPO (eosinophil peroxidase) is the most abundant granular protein and catalyzes the reaction of H2O2 with thiocyanate or bromide to generate oxidants that aim to oxidize the cysteine of thiol groups that are abundant in mucin polymers, basically MUC5AC with modest increase of MUC5B [7]. In turn, in allergic inflammation, two signal systems are activated in the airway cells, IL-13/4 and EGFR. IL-13 activates STAT (signal transducer and activator transcription), which induces mucin production in Clara cells. Endogenous ATP stimulates the P2Y2 receptor on the apical surface of secretory cells, the second messenger is activated, and the mucin polymers are released into the lumen forming a stiff mesh of very compact mucus [47]. High levels of endobronchial mucus inversely correlate with FEV1, basically by occluding subsegmental airways causing gas trapping. In fact, the mucus impaction of the distal routes can be carried out with minimal sputum expectoration.
When a foreign element invades the airway (for example Rhinovirus) NF-kβ is activated, which stimulates MMP which releases EGFR and this induces the release of MUC5AC [48]. Already, the genes that encode the secretion of mucins have been cloned, allowing their investigation as an essential component of normal and pathological mucus. Mucus impaction in small pathways (a relatively silent area of the airway) is not easy to identify clinically or physiologically, either by radiological or endoscopic examinations. In fact, only between 20-40% of asthmatic patients is detected mucus hypersecretion, being even more viscous than that of patients with COPD and colloidal appearance (Figure 3). Mucoid impaction in small airways cannot necessarily be seen on HCRT (high resolution computed tomography) images. Yoshida and collaborators have used a CT technique, called MPR (curved multiplanar reconstruction) that allows the visualization of airway images longitudinally. This analysis makes it possible to quantitatively measure the impacted mucus. Airways <2 mm (small airways) include the 8-23rd generation. The fourth and fifth generations seem to be the most affected by impaction in bronchial asthma. This could have therapeutic implications in the future [49].
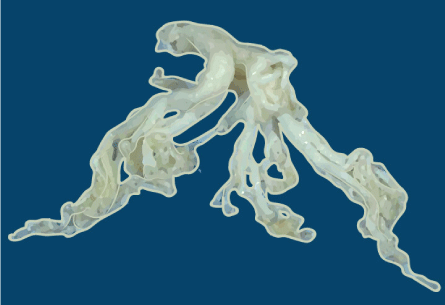
Figure 3. Mucus plug
The cast of the bronchial tree is formed of inspissated mucus and was coughed up by a patient during an asthmatic attack.
Charcot-Leyden crystals deserve separate mention because they could be future therapeutic targets in asthma. These needle-shaped crystals (10-40 µ) are associated with the presence of eosinophils, and can be observed in the sputum of asthmatic patients. Described in 1853 by Charcot, it was originally believed that they were formed by spermine and mucin as precursor [50]. Later it was postulated that it was formed by the phospholipase of the eosinophils membrane [51]. We currently know that the constituent protein is galecine-10 [52]. Galectins are proteins that bind glycan or lectin with affinity for beta-galactose-modified glycol-conjugates that is they have carbohydrates recognition domain. Twenty human galectins generated by the splicing of transcribed messenger pre-RNAs of 11 are known. Only galectin-10 crystallizes rapidly and spontaneously under certain conditions, including exocytosis of eosinophils into the extracellular environment. Apparently, the Tyrosine69 polar residue is vital for crystallization [53]. Studies in mice have shown that antibodies against protein dissolve crystals [54]. Dissolution of the crystals could decrease elasticity and improve clearance of impacted mucus in patients with eosinophilic asthma and chronic rhino-sinusitis with polyposis. However, crystals may also come from basophils and medications for clinical use yet not available in human.
In addition to the mucolytic effect, the dissolution of the crystals can have anti-inflammatory effects by blocking the activation of the innate and acquired immune response induced by the crystals. Studies in members of the family camelidae (camels and llamas) with inhaled nanobodies (therapeutic proteins with maximum antibody function, which only have heavy chains), have demonstrated certain suitable characteristic such as small size, thermal stability, high solubility and systemic half-life short [7]. Obviously, future studies in humans are required. Crystal research could help underpin type-2 immunity [55]. Curschchmann' spirals are another morphological expression of mucus impacted in the sputum of patients with asthma (Figure 4) [56].
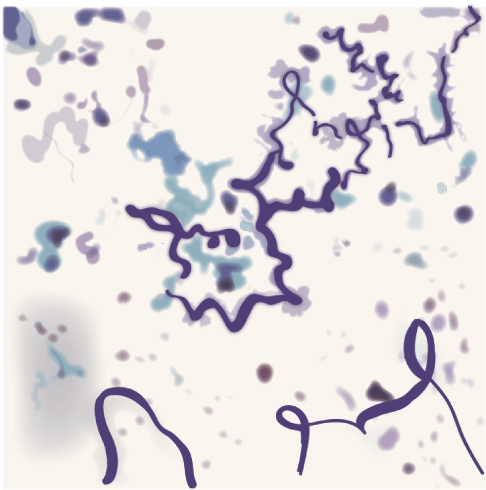
Figure 4. Curschmann’ spirals
Curschmann’ spirals can be seen on a wet preparation or on a Papanicolaou-stained specimen of respiratory secretions. They are associated with the production of excess mucus (asthma, bronchitis, and smoking).
Based on epidemiological studies, the global prevalence of COPD is estimated at 11.7%, with 384 million affected and 3 million death per year (6% of all deaths), becoming a leading cause of morbidity and mortality [57,58]. With the increase in smoking in developing countries and the increase in life expectancy in high-income countries, it is expected that the prevalence will increase in the next 30 years and it is like that in the year 2030 there will be a total of annual deaths higher than 4.5 million [59]. In these evaluations there is a sub-registry, since most of the studies are carried out smoking patients and nowadays the prevalence of COPD in non-smokers is increasing, associated with the use of biomass, other fuels and environmental pollution [60].
COPD manifests as persistent airflow obstruction, most often in response to inhaled environmental agents [61]. Hyperconcentrated mucus has been reported in lower-airway samples obtained from patients with COPD, and higher mucus concentrations are correlated with reduced in vivo rates of mucociliary clearance and cigarette smoke, is associated with an increase in mucin concentration [62]. Exposure to cigarette smoke may produce abnormalities in CFTR-mediated secretion of chloride anions through oxidant-induced reduction of CFTR transcription rates and direct damage to CFTR protein in the apical membrane [63,64]. These defects in epithelial ion and fluid transport (hydration) are amplified by cigarette smoke–induced hypersecretion of MUC5AC and MUC5B [65].
The important role of airway mucus hypersecretion in COPD should be fully recognized [66]. Airway mucus hypersecretion, via chronic cough and expectoration, reduces airflow and exercise capacity and increases the risk of acute exacerbation, mortality, and poor prognosis in COPD patients [67].
Non-cystic bronchiectasis
Non–cystic fibrosis bronchiectasis is a phenotype defined by bronchiectasis in the absence of a nogenetic cause [68]. Bronchiectasis is bronchial deformation and persistent bronchial expansion caused by the destruction of smooth muscles and elastic tissues on the bronchial wall due to chronic purulent inflammation and fibrosis of the bronchus and surrounding lung tissues [69]. Cough occurs in >90% of patients with bronchiectasis, and expectoration occur as a complication in 75%–100% of patients with cough. Patients with bronchiectasis show reduced mucociliary activities in the airway and reduced expectoration ability, and the biophysical properties of their mucus are altered, eg, it is more viscous [70]. The long-term accumulation of mucus in the airway facilitates colonization by bacteria as well as recurrent cough and expectoration [70]. In this way, airway mucus hypersecretion is a fundamental pathophysiological and clinical feature of bronchiectasis. Linked with airway mucus hypersecretion, airway inflammation and damage occur in bronchiectasis patients, seriously affecting patients’ quality of life. The true incidence remains unknown in many populations. The prevalence increases with age. The general belief is that the incidence is decreasing. This is attributed to the introduction of antibiotics and immunization for children and for these reasons is not considered a major health problem and some consider it an “orphan disease”. Although non–cystic fibrosis bronchiectasis is now typically defined by dilated bronchi on CT, classic pathological studies have highlighted severe small-airway disease, including bronchiolectasis, mucus plugging, and inflammation, and bronchiectasis has the typical small-airway airflow impairment of muco-obstructive diseases [71]. Currently, genetic studies have not linked ion-transport genes to non–cystic fibrosis bronchiectasis [72]. Anaerobic bacteria, staphylococcus, and H. influenzae, with approximately 15 to 20% of patients positive for P. aeruginosa are the most frequent bacteria in this context [73,74]. Regardless of the underlying cause, this chronic airway bacterial infection (which is what happens in bronchiectasis) active and recruit phagocytic cells and generates oxidative stress in a scenario where there is a dysregulation of the innate and adaptive immune response. Cytokines and their receptors may be therapeutics targets [71].
The interrelationship between the three entities (COPD, bronchial asthma and bronchiectasis) is complex and unclear. The prevalence of obstructive diseases in general is increasing in recent decades and increase with age. In the beginning bronchiectasis they are also an obstructive phenomenon probably begins in the small airway [75]. With the evolution of the disease, disorders restrictive added due to the progressive destruction of parenchyma lung [76]. Statistically powerful and excluding Allergic Bronchopulmonary Aspergillosis (ABPA) studies have found varicose bronchiectasis in 60% of patents with non-severe allergic asthma and 50% of severe asthmatic using HRCT scan [77]. Unlike APBA can affect all lobes and both proximal and distal airway. Therefore the recommendation of the guidelines is that asthma in adults should be considered an etiology of bronchiectasis if another can’t be identified [8]. However, there is no robust evidence that COPD is cause of bronchiectasis. This relationship requires further studies [78]. What does seem clear is that the three are inflammatory conditions that cause oxidative stress that damage cells and subcellular compartments.
Cystic fibrosis
In 1989 the gene encoding cystic fibrosis was identified on the long arm of chromosome 7. It is a large gene that contains more than 250,000 bases with 27 exons, and that encodes a 1,480 amino acid protein, called the cystic fibrosis transmembrane conductance regulator (CFTR), this protein is clearly a chlorine channel activated by cyclic AMP. It is the genetic disease that leads to higher mortality in white people (the most frequent mutation is the Delta-F 508 deletion). The normal channel is responsible for producing sweat, digestive juices, and various mucus fluids [67]. Patients with pulmonary cystic fibrosis show severe airway mucus hypersecretion, and many suffer recurrent pulmonary infections, which can accelerate decline in the lung function. Patient sputum is characterized by obvious neutrophil infiltration, cellular DNA released from injured cells, and abundant Pseudomonas aeruginosa [79]. Patients also show substantial epithelial goblet cell metaplasia in the airway in contrast to healthy individuals, as well as significantly higher MUC5AC expression [79]. Airway mucus hypersecretion in patients with pulmonary cystic fibrosis is associated with persistent cough, expectoration, and dyspnea. Laboratory evidence suggests that cystic fibrosis can be triggered by viral infections that lead to unrestrained liquid absorption, mucus hyperconcentration, and the formation of mucus plaques and plugs [80]. Sputum measurements revealed higher total mucin concentrations and a higher percentage of solids patients with cystic fibrosis than in healthy persons [20]. Recent data regarding bronchoalveolar lavage from children with cystic fibrosis showed that a persistent increase in mucin concentration and inflammatory cells preceded bacterial infection in the lung [81]. Aspirated oral anaerobes appear to be the first bacterial pathogens in the lungs of patients with cystic fibrosis, followed by the classic gram-negative pathogens that probably accelerate the vicious cycle depicted in and loss of lung function [82,83].
Primary Ciliary Dyskinesia
This syndrome was initially recognized based on the triad of chronic sinusitis, bronchiectasis, and situs inversus (Kartagener syndrome) [84] and Afzelius later recognized that these patients had “immotile” cilia and defective ciliary ultrastructure [85]. Over time, it was recognized that most patients had stiff, uncoordinated, and/or ineffective ciliary beat, and “primary ciliary dyskinesia” was used to distinguish this ciliary genetic disorder from secondary or acquired ciliary defects. Even though Primary Ciliary Dyskinesia (PCD) has an estimated incidence of 1 per 10,000–20,000 births, based on population surveys of situs inversus and bronchiectasis, it is difficult to determine the prevalence of PCD in the world, largely due to sub-optimal diagnostic approaches [86]. Cilia are evolutionarily conserved organelles and motile respiratory cilia have a complex (9 + 2) axonemal structure to generate functional ciliary motility [87,88]. Nexin–dynein regulatory complexes (“nexin links”) connect the doublets, and radial spokes connect the doublets to the central pair for structural support during cilia bending [89]. Mutations in genes necessary for the biogenesis of cilia, or genes encoding the axonemal structure and/or functional components of motile cilia, can result in PCD. During early development, each cell in the embryonic ventral node contains a single motile cilium. This specialized cilium has 9 peripheral doublets and dynein arms, but lacks the central pair of microtubules (9 + 0 axonemal structure) [90]. Functionally, this cilium has a rotary motion, which drives a vectorial movement and laterality of organ lateralization during embryogenesis. When nodal ciliary function is absent, organ lateralization is random.
The pathogenesis of PCD is not only a motor problem caused by genetic abnormalities in cilial shaft proteins, cilial beat frequency, and defective mucociliary clearance (Figure 5) [91]. Sputum obtained from patients with PCD also is abnormally hyperconcentrated, contributing to the pathogenesis of the disease [92]. Ciliary sensitivity, and not just the motor component, is defective contributing thereby decrease of fluid secretion and generating a hyperconcentrated mucus plaque or plug component to airflow obstruction. The typical clinical phenotype in PCD includes (1) neonatal respiratory distress, and/or (2) chronic, persistent lower respiratory symptoms (early onset and persistent wet cough), and/or (3) chronic, persistent upper respiratory symptoms (nasal congestion and otitis media), and/or (4) a laterality defect (situs inversus or ambiguus). Indeed, the presence of any two of these four hallmark clinical features provides a strong clinical phenotype for PCD, assuming that CF has been excluded [93]. For adults, all males with abnormal spermatozoal movement should be evaluated for PCD, if they have respiratory symptoms. Several medical disorders and phenotypes may coexist with PCD, including complex congenital heart disease, laterality defects, retinitis pigmentosa, hydrocephalus, pectus excavatum, and scoliosis (4).
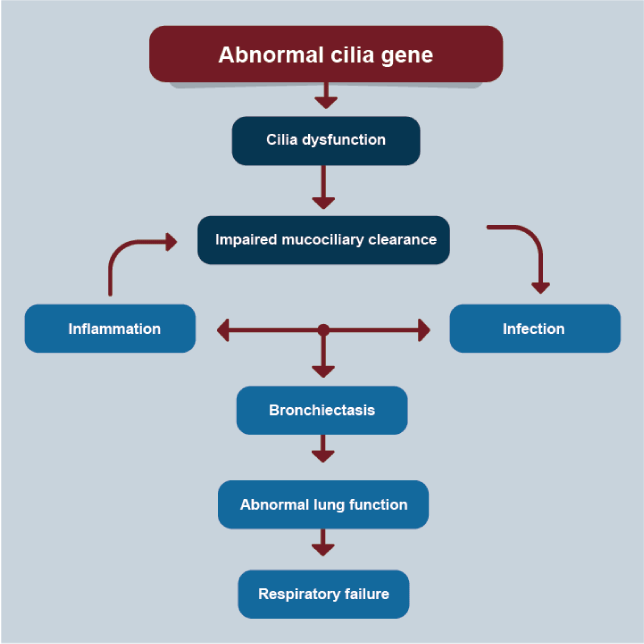
Figure 5. PCD
Proposed pathogenesis of primary ciliary dyskinesia lung disease.
Airway mucus hypersecretion manifests mainly as chronic cough and expectoration, which are particularly obvious during stimulation, weather changes, and exacerbation of infection. The severity of cough and the volume and properties of sputum serve as an index of airway mucus hypersecretion, which can fluctuate during periods of stability or acute exacerbation of airway diseases. The cough and sputum assessment questionnaire (CASA-Q), St George’s Respiratory Questionnaire and American Thoracic Society Questionnaire, are important questionnaires that are used for many cohorts around the world and also contain questions for evaluation of cough and sputum, can also be used to evaluate airway mucus hypersecretion [94-96]. The tools are used to make a general diagnosis of muco-obstructive disease.
Disease-specific criteria assist in the diagnosis of each muco-obstructive lung disease: for cystic fibrosis, levels of chloride anions in sweat and genetic testing [97]; for COPD, exposure history and spirometry (2); for bronchial asthma clinical history, spirometry, peripheral cell count and sputum and immunological studies [98], PCD, nasal nitric-oxide measurements, cilia waveform analyses, and genetic testing [4,91]; and for non–cystic fibrosis bronchiectasis, CT scanning (68). Cytokine and chemokine measurements in sputum aid in establishing the inflammatory component of airway muco-obstruction. Measures of mucus concentration (percent solids) and total mucin concentrations have also been explored as diagnostic tools and mucin concentration is a good biomarker for COPD [13].
Given the important clinical significance of airway mucus hypersecretion in chronic airway inflammatory diseases, therapy is widely used to help relieve airway stenosis, avoid recurrent infection and exacerbation, and delay decline in lung function. At present, chronic airway inflammatory diseases such as COPD and asthma are treated mainly using bronchodilators to open occluded airways or by using corticosteroid inhalation to control inflammation [99]. Blocking type 2 immunity with monoclonal antibodies is a strategy for the treatment of refractory eosinophilic asthma. Anti-IL-5 antibodies have been used in COPD with eosinophilic phenotype with a reduction in exacerbations, but not in symptoms or quality of life [100].
A direct approach to treatment of muco-obstructive lung diseases is to reduce the concentration of pathologic mucus — that is, rehydrate it, with inhalation of osmotically active aerosols (e.g., hypertonic saline or mannitol) [101,102]. Both treatments modulate the ion transport that may redirect airway epithelial ion transport from net absorptive to secretory directions, providing a mechanism for epithelial restoration of airway-surface hydration [103]. Hypertonic saline helps clear sputum, and administering 7% hypertonic saline to patients with pulmonary cystic fibrosis accelerates mucus discharge and improves airflow and lung function [104]. Aerosol inhalation of 7% hypertonic saline as an auxiliary therapy can promote mucus discharge and improve lung function and quality of life in patients with bronchiectasis. It has also been used in PCD, with some early evidence of efficacy [105]. Bronchial asthma has no indication for this therapy. Some COPD patients develop dyspnea when receiving atomizing hypertonic saline. Therefore, hypertonic saline treatment should be used in accordance with its clinical indications [106]. Anderson and collaborators have developed a spray dried preparation of mannitol and found that bronchial responsiveness to inhaling mannitol identified people with currently active asthma, and mannitol had potential to replace the ‘osmotic’ benefits of exercise and could be used as a treatment to enhance mucociliary clearance in patients with cystic fibrosis [101].
N-acetylcysteine (NAC) has direct and indirect properties as an antioxidant in the treatment of COPD. NAC free-thiol is capable of interacting with electrophilic groups of ROS (reactive oxidant species). Indirectly, NAC is a precursor of GHS (glutathione), a neutralizer of ROS. An Israeli study found beneficial effects of NAC in the gas trap [107]. The HIACE study (1200 mg/day oral NAC for one year), showed significant improvement in the function of the small airways, and the frequency of exacerbations [108]. Antioxidant mucolytics are recommended only in selected patients in COPD, according to GOLD 2020 [109]. Little evidence exists for or against the use of N-acetylcysteine in patients with asthma, pulmonary cystic fibrosis, bronchiectasis or PCD; large, rigorous trials are needed to examine safety and efficacy in these populations. Inhaled acetylcysteine has not proved to have the mucin-reductive activity required for a therapeutic effect [14].
Disease-specific therapies for one muco-obstructive disease, cystic fibrosis, are now available. Ivacaftor (VX-770) is a potentiator of residual CFTR function that has been approved for patients with cystic fibrosis with gating and some splicing CFTR mutations [110]. For patients with cystic fibrosis who are homozygous for a deletion of phenylalanine 508 (F508del) in CFTR, ivacaftor–lumacaftor and tezacaftor–ivacaftor are available but offer more modest clinical benefit. Three-drug combinations of correctors and potentiators appear to be highly effective in patients with at least one F508del mutation (approximately 90% of patients).
Airway mucus hypersecretion is an important driver of airway obstruction, rapid decline in lung function, and increased frequency of acute exacerbation in patients with chronic airway inflammatory diseases; it is closely related to patient prognosis.
Muco-obstructive diseases are characterized by mucus hyperconcentration. The five muco-obstructive diseases differ with respect to the epithelial abnormalities that produce mucus hyperconcentration but follow a final common path of mucus concentration–dependent formation of mucus plaques and plugs.
Detailed investigation into the signaling pathways and downstream effector molecules involved in airway mucus hypersecretion will improve our understanding and the treatment of these chronic diseases.
Therapies that are designed to rehydrate and restore mucous viscous or elastic properties are rational. The challenge is to deliver these therapies to the small airways, where mucus obstruction may be complete and the physics of aerosol-deposition efficiency may be poor.
Clinicians should recognize the benefits of expectorant therapies for patients with chronic airway inflammatory diseases and should continuously seek to improve and innovate treatments for such patients.
No.
No.
This work was only carried out by the author. Author AA contributed in the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
- Button B, Goodell HP, Atieh E, Atieh E, Chen YC, et al (2018). Roles of mucus adhesion and cohesion in cough clearance. Proc Natl Acad Sci USA 115: 12501-12506.
- Han MK, Agusti A, Calverley PM, Celli BR, Criner G, et al. (2010) Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 182: 598-604. [Crossref]
- King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW (2006) Characterization of the onset and presenting clinical features of adult bronchiectasis. Respir Med 100: 2183-2189. [Crossref]
- Knowles MR, Zariwala M, Leigh M (2016) Primary ciliary dyskinesia. Clin Chest Med 37: 449-461. [Crossref]
- Rowe SM, Miller S, Sorscher EJ (2005) Cystic fibrosis. N Engl J Med 352: 1992-2001.
- Capistrano SJ, van Reyk D, Chen H (2017) Evidence of biomass smoke exposure as a causative factor for the development of COPD. Toxics 5: 36. [Crossref]
- Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler SM, et al (2018) Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest 128: 997-1009. [Crossref]
- Pasteur MG, Bilton D, Hill AT (2010) British Thoracic Society guidelines for non-CF bronchiectasis. Thorax 65 Suppl 1: 1-58. [Crossref]
- King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW (2006) Characterization of the onset and presenting clinical features of adult bronchiectasis. Respir Med 100: 2183-2189. [Crossref]
- Rowe SM, Miller S, Sorscher EJ (2005) Cystic fibrosis. N Engl J Med 352: 1992-2001.
- Knowles MR, Zariwala M, Leigh M (2016) Primary ciliary dyskinesia. Clin Chest Med 37: 449-461. [Crossref]
- Boucher RC (2019) Muco-obstructive lung diseases. N Engl J Med 380: 1941-1953. [Crossref]
- Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, et al (2017) Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med 377: 911-922. [Crossref]
- Fahy JV, Dickey BF (2010) Airway mucus function and dysfunction. N Engl J Med 363: 2233-2247. [Crossref]
- Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, et al (2019) Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med 199: 715-727. [Crossref]
- Widdicombe JH, Wine JJ (2015) Airway gland structure and function. Physiol Rev 95: 1241-1319.
- Button B, Cai LH, Ehre C, Kesimer M, Hill DB, et al (2012) A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337: 937-941. [Crossref]
- Thornton DJ, Rousseau K, McGuckin MA (2008) Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol 70: 459-486. [Crossref]
- Woodruff PG, Barr RG, Bleecker E, Christenson SA, Couper D, et al (2016) Clinical significance of symptoms in smokers with preserved pulmonary function. N Engl J Med 374: 1811-1821.
- Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, et al (2014) Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest 124: 3047-3060. [Crossref]
- Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, et al (2002) Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol 169: 6985-6991. [Crossref]
- Cole AM, Dewan P, Ganz T (1999) Innate antimicrobial activity of nasal secretions. Infect Immun. 67: 3267-3275. [Crossref]
- Livraghi-Butrico A, Grubb BR, Wilkinson KJ, Volmer AS, Burns KA, et al (2017) Contribution of mucus concentration and secreted mucins Muc5ac and Muc5b to the pathogenesis of muco-obstructive lung disease. Mucosal Immunol 10: 395-407. [Crossref]
- Chalmers JD, Moffitt KL, Suarez-Cuartin G, Sibila O, Finch S, et al (2017) Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am J Respir Crit Care Med 195:1384-1393. [Crossref]
- Sly PD, Gangell CL, Chen L, Ware RS, Rauganathan S, et al (2013) Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 368: 1963-1970. [Crossref]
- Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, et al (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109: 317-325. [Crossref]
- Gleeson K, Eggli DF, Maxwell SL (1997) Quantitative aspiration during sleep in normal subjects. Chest 111: 1266–1272. [Crossref]
- Flynn JM, Niccum D, Dunitz JM, Hunter RC (2016) Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog 12: e1005846-e1005846. [Crossref]
- Tunney MM, Field TR, Moriarty TF, Patrick S, Doeringer G, et al (2008) Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 177: 995-1001. [Crossref]
- Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers T, et al (2006) A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci U S A 103: 18131-18136. [Crossref]
- Sze MA, Hogg JC, Sin DD (2014) Bacterial microbiome of lungs in COPD. Int J Chron Obstruct Pulmon Dis 9: 229-238. [Crossref]
- Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, et al (2010) A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS 14: 9-59. [Crossref]
- Sze MA, Dimitriu PA, Hayashi S, Elliot WM, McDorough JE, et al (2012) The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 185: 1073-1080. [Crossref]
- Rosenfeld M, Emerson J, Williams-Warren J, Pepe M, Smith A, et al (2001) Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr 139: 359-365. [Crossref]
- Sethi S, Maloney J, Grove L, Wrona C, Berenson CS (2006) Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 173: 991-998. [Crossref]
- Goss CH, Burns JL (2007) Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax 62: 360-367. [Crossref]
- Boucher RC (2015) On the pathogenesis of acute exacerbations of mucoobstructive lung diseases. Ann Am Thorac Soc 12 (Suppl 2): S160-S163. [Crossref]
- Lee AL, Button BM, Denehy L, Roberts SJ, Bamford TL, et al (2014) Proximal and distal gastro-oesophageal reflux in chronic obstructive pulmonary disease and bronchiectasis. Respirology 19: 211-217. [Crossref]
- Brodlie M, Aseeri A, Lordan JL, Robertson AGN, McKean MC, et al (2015) Bile acid aspiration in people with cystic fibrosis before and after lung transplantation. Eur Respir J 46: 1820-1823. [Crossref]
- Cox MJ, Turek EM, Hennessy C, Mirza GK, James PL, et al (2017) Longitudinal assessment of sputum microbiome by sequencing of the 16S rRNA gene in non-cystic fibrosis bronchiectasis patients. PLoS One 12: e0170622-e0170622. [Crossref]
- Curran DR, Cohn L (2010) Advances in mucous cell metaplasia: a plug for mucus as a therapeutic focus in chronic airway disease. Am J Respir Cell Mol Biol 42: 268–275. [Crossref]
- Cerveri I, Brusasco V (2010) Revisited role for mucus hypersecretion in the pathogenesis of COPD. Eur Respir Rev 19: 109-112.
- Dijkstra AE, Boezen HM, van den Berge M, Von JM, Hiemstra PS, et al (2015) Dissecting the genetics of chronic mucus hypersecretion in smokers with and without COPD. Eur Respir J 2015: 60-75.
- Barnes PJ (2009) Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183-192. [Crossref]
- To T, Stanojevic S, Moores G, Gerson AS, Bateman ED, et al. (2012) Global asthma prevalence in adults: finding from the cross-sectional World Health Survey. BMC Public Health 12: 204. [Crossref]
- Hogg JC, Hegel PG (1997) Postmorten pathology. In: Barnes PJ, Grustein MM, Leff AR, Woolcook AJ (Eds) Asthma Lippincott-Raven. New York. 201-208.
- Evans CM, Kim K, Tuvim MJ, Dickey BF (2009) Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med 15: 4-11. [Crossref]
- Burgel PR, Nadel JA (2010) Plugging (publicizing) to prevent mucous plugging. Eur Respir J 36: 1236-1238.
- Yoshida Y, Takaku Y, Nakamoto Y, Takayani N, Yanagisawa T, et al (2020) Changes in airway diameter and mucus plugs in patients with asthma exacerbation. PLoS One. 15(2): e0229238. [Crossref]
- Charcot JM, Robin C (1853) Observation de leucocythémie. Cir SG (Eds) Soc Biol 5: 44.
- Guido I, Alvarado A (1991) La reacción tardía en asma bronquial. AMC 34: 94-111.
- Su J (2018) A breif history of Charcot-Leyden protein/galectin-10 research. Molecules 23: 2931. [Crossref]
- Cummings RD, Liu FT, Vasta GR (2017) Galectins. In: Varki A, Cummings RD, Esko JD (Eds) Esentials of glycobiology. (3rd Edn) Cold Spring Harborg, Cold Spring Harbor laboratory Press, NY. [Crossref]
- Person EK, Verstraete K, Heyndrickx I, Gevaert E, Aegaerter H, et al (2019) Protein crystallization promotes type2 immunity and is reversible by antibody treatment. Science 24: 364-364.
- Fahy JV, Locksley RM (2019) Making Asthma Crystal Clear. N Engl J Med 381: 882-884. [Crossref]
- Vezza PR, Montgomery EA (1998) Curschmann’ spirals. N Engl J Med 338: 1043.
- Adeloye D, Chua S, Lee C, Basquill C, Papana A, et al. (2015) Global and regional estimates of COPD prevalence: systematic review and meta-analysis. Glob Health 5: 020415. [Crossref]
- Global Burden of Diseases Study Collaborators (2015) Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Diseases Study 2013. Lancet 385: 117-171.
- WHO (2015) Projection of mortality and causes of death, 2015 and 2030. World Health Organization.
- Alvarado A (2018) Chronic obstructive pulmonary disease in non-smokers: An update. Clin Res Trials 4: 1-8.
- Anderson WH, Coakley RD, Button B, Henderson AG, Zeman KL, et al. (2015) The relationship of mucus concentration (hydration) to mucus osmotic pressure and transport in chronic bronchitis. Am J Respir Crit Care Med 192:182-190. [Crossref]
- Hogg JC, Paré PD, Hackett TL (2017) The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol Rev 97: 529-552. [Crossref]
- Clunes LA, Davies CM, Coakley RD, Aleksandrov AA, Henderson AG, et al. (2012) Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB 26: 533-545. [Crossref]
- Cantin AM, Bilodeau G, Ouellet C, Liao J, Hanrahan JW (2006) Oxidant stress suppresses CFTR expression. Am J Physiol Cell Physiol 290: C262-C270. [Crossref]
- Rogers DF (2007) Physiology of airway mucus secretion and pathophysiology of hypersecretion. Respir Care 52: 1134-1146. [Crossref]
- Miravitlles M (2011) Cough and sputum production as risk factors for poor outcomes in patients with COPD. Respir Med 105: 1118-1128. [Crossref]
- Shen Y, Huang S, Kang J, Lin J, Lai K, et al. (2018) Management of airway mucus by hypersecretion in chronic airway inflammatory disease: Chinese expert consensus (English edition). Int J Chron Obstruct Pulmon dis 13: 399-402. [Crossref]
- Flume PA, Chalmers JD, Olivier KN (2018) Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet 392: 880-890. [Crossref]
- Zhonghua J, He H, Hu X, Za Z (2012) Workgroup of bronchiectasis diagnosis and management Expert consensus on diagnosis and management of adult bronchiectasis. 35: 485-492.
- Tambascio J, de Souza HC, Martinez JA, Afonso JL, Jardim JR, et al. (2013) The influence of purulence on ciliary and cough transport in bronchiectasis. Respir Care 58: 2101-2106.
- Attenburg J, Wortel K, Van dr Werf TS, Boersnm WG (2015) Non-cystic fibrosis bronchiectasis: clinical presentation, diagnosis, and treatment, illustrated by data from Dutch Teaching Hospital. MJM 73:147-154. [Crossref]
- Bienvenu T, Sermet-Gaudelus I, Burgel PR, Hubert D, Crestani B, et al. (2010) Cystic fibrosis transmembrane conductance regulator channel dysfunction in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 181: 1078-1084.
- Brodlie M, Aseeri A, Lordan JL, Andrew GN, McKean MC, et al. (2015) Bile acid aspiration in people with cystic fibrosis before and after lung transplantation. Eur Respir J 46: 1820-1823. [Crossref]
- Serisier DJ, Martin ML, McGuckin MA, Lourie R, Chen AC, et al. (2013) Effect of long-term, low-dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA 309: 1260-1267. [Crossref]
- Athanazio R (2012) Airway disease: similarities and differences between asthma, COPD and bronchiectasis. Clinics (Sao Paulo) 67: 1335-1343. [Crossref]
- Hamutcu R, Rowland JM, Horn MV, Kaminsky C, MacLaughlin EF, et al. (2002) Clinical findings and lung pathology in children with cystic fibrosis. Am J Respir Crit Care Med 165: 1172-1175. [Crossref]
- Paganin F, Séneterre E, Chanez P, Daurés JP, Bruel JM, et al. (1996) Computed tomography of the lungs in asthma: influence of disease severity and etiology. Am J Respir Crit Care Med 153: 110-114. [Crossref]
- NICE (NIFfCE) (2004) Chronic obstructive pulmonary disease: national clinical guidelines for management of chronic obstructive pulmonary disease in adult in primary and secondary care. Thorax 59: 1-232. [Crossref]
- Kreda SM, Davis CW, Rose MC (2012) CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb Perspect Med 2: a009589. [Crossref]
- Tarran R, Button B, Picher M, Paradiso AM, Ribeiro CM, et al. (2005) Normal and cystic fibrosis airway surface liquid homeostasis: the effects of phasic shear stress and viral infections. J Biol Chem 280: 35751-35759. [Crossref]
- Esther CR, Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, et al. (2019) Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med 11: eaav3488-eaav3488.
- Abdullah LH, Coakley R, Webster MJ, Shu Y, Tarran R, et al. (2018) Mucin production and hydration responses to mucopurulent materials in normal versus cystic fibrosis airway epithelia. Am J Respir Crit Care Med 197: 481-491. [Crossref]
- Muhlebach MS, Zorn BT, Esther CR, Hatch JE, Murray CP, et al. (2018) Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog 14: e1006798-e1006798. [Crossref]
- Kartagener M (1933) Zur pathogenese der bronkiectasien: bronkiectasien bei situs viscerum inversus. Beitr Klin Tuberk 82: 489-501.
- Afzelius BA (1976) A human syndrome caused by immotile cilia. Science 193: 317-319. [Crossref]
- Lucas JS, Leigh MW (2014) Diagnosis of primary ciliary dyskinesia: searching for a gold standard. Eur Respir J 44: 1418-1422.
- Salathe M (2007) Regulation of mammalian ciliary beating. Annu Rev Physiol 69: 401-422. [Crossref]
- Satir P, Christensen ST (2007) Overview of structure and function of mammalian cilia. Annu Rev Physiol 69: 377-400. [Crossref]
- Chilvers MA, Rutman A, O'Callaghan C (2003) Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol 112: 518-524. [Crossref]
- Basu B, Brueckner M (2008) Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr Top Dev Biol 85: 151-174. [Crossref]
- Horani A, Ferkol TW, Dutcher SK, Brody SL (2016) Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev 18: 18-24. [Crossref]
- Bush A, Payne D, Pike S, Jenkins G, Henke MO, et al. (2006) Mucus properties in children with primary ciliary dyskinesia: comparison with cystic fibrosis. Chest 129: 118-123.
- Sagel SD, Davis SD, Campisi P, Dell SD (2011) Update of respiratory tract disease in children with primary ciliary dyskinesia. Proc Am Thorac Soc 8: 438-443. [Crossref]
- Crawford B, Monz B, Hohlfeld J, Roche N, Rubin B, et al. (2008) Development and validation of a cough and sputum assessment questionnaire. Respir Med 102: 1545-1555. [Crossref]
- Monz BU, Sachs P, McDonald J, Crawford B, Nivens MC, et al. (2010) Responsiveness of the cough and sputum assessment questionnaire in exacerbations of COPD and chronic bronchitis. Respir Med 104: 534-541.
- Hardin M, Rennard SI (2017) What’s new with the St George’s Respiratory Questionnaire and why do we care? Chronic Obstr Pulm Dis 4: 83-86. [Crossref]
- Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, et al. (2017) Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr 181S: S4-S15.e1 [Crossref]
- Alvarado A (2019) Differences, similarities and controversies between bronchial asthma and chronic obstructive pulmonary disease. Clin Res Trials 5: 1-12.
- GINA (2019) Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma.
- Criner GJ, Celli BR, Brightling CE, Augusti A, Papi A, et al. (2019) Benralizumab for Prevention of COPD Exacerbations. N Engl J Med 1023-1034. [Crossref]
- Anderson SD, Daviskas E, Brannan JD, Chan HK (2018) Repurposing excipients as active inhalation agents: the mannitol story. Adv Drug Deliv Rev 133: 45-56. [Crossref]
- Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, et al. (2006) Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med 354: 241-250.
- Shei RJ, Peabody JE, Kaza N, Rowe SM (2018) The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr Opin Pharmacol 43: 152-165. [Crossref]
- Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, et al. (2006) A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 354: 229-240. [Crossref]
- Paff T, Daniels JM, Weersink EJ, Lutter R, Vonk Noordegraaf A, et al. (2017) A randomized controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. Eur Respir J 49: 1601770. [Crossref]
- Fujimoto K, Yasuo M, Urushibata K, Hanaoka M, Koizumi T, et al. (2005) Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. Eur Respir J 25: 640-646. [Crossref]
- Stav HN, Raz M (2009) Effect of N-acetylcysteine on air trapping in COPD: a randomized placebo-controlled study. Chest 136: 381-386. [Crossref]
- Tse HN, Raiteri L, Wong KY, Yee KS, Ng LY, et al. (2013) High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled. HIACE study. Chest 144: 106-118. [Crossref]
- GOLD (2020) Global Strategy for the Diagnosis, Management and Prevention of COPD. Global Initiative for Chronic Obstructive Lung Diseases.
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, et al. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365: 1663-1672. [Crossref]