Background: Anti-apolipoprotein A-1 antibodies (anti-apoA-1 IgGs) were raised in different diseases associated with increased risk of cardiovascular events (CV), including HIV-infected individuals. In humans, anti-apoA-1 antibody response is directed toward the C-terminal alpha helix of apoA-1 (cterA1). CterA1 mimetic peptides could be used to detect anti-apoA-1 IgGs and inhibit anti-apoA-1 IgGs pro-inflammatory effects.
Aim: To assess the prevalence of anti-cterA1 IgGs in HIV patients enrolled in the Swiss cohort, their associations with biomarkers of HIV infection severity and inflammation, the ability of cterA1 mimetic peptides to reverse the HIV-derived ssRNA40-induced foam cell formation of macrophages.
Result: 26% of HIV patients were positive for anti-cterA1 IgGs. Anti-cterA1 IgG seropositivity was independently associated with higher viremia (adjusted odds ratio [aOR]: 5.02; 95% Confidence Interval [95%CI]:2.41-16.9; p=0.0029), and lower CD4+count (aOR: 2.57;95%CI:1.10-6.03; p=0.029). Anti-cterA1 IgG seropositive individuals had lower total and LDL cholesterol values (p<0.03) and increased levels of TNF-α, IL-8 and IL-10 (p<0.008). The cterA1 mimetic peptide was found to revert the ssRNA40-induced foam cell formation.
Conclusion: High levels of anti-cterA1 IgGs are associated with biological HIV disease severity and with a pro-inflammatory profile. By reversing the ssRNA40-induced foam cell formation, cterA1 mimetic peptide may represent an anti-atherosclerotic therapeutic agent in HIV settings.
HIV, Anti-apoA-1 IgG, Inflammation, CterA1 peptide, Macrophage, Foam cells
HIV-infected individuals exhibit an increased risk of atherosclerosis and cardiovascular disease (CVD), leading to a higher morbidity and mortality rate in this population [1,2]. In addition to a high prevalence of traditional CV risk factors among HIV-infected individuals, specific factors linked to HIV infection have also been shown to accelerate the development of atherosclerosis and its associated CV complications. These include i) persistent immune-driven inflammation characterized by an increased pro-inflammatory cytokine profile, macrophage activation and endothelial dysfunction, ii) HIV or anti-retroviral therapy (ART)-related factors, such as high viremia and CD4+ lymphopenia or dyslipidemia and insulin resistance, respectively [3-6].
Imbalanced immune system activation following HIV infection results in the production of a wide range of autoantibodies as a consequence of uncontrolled B-cell proliferation and molecular mimicry between HIV proteins and self-antigens [7-10]. Among autoantibodies of possible CV clinical relevance, antibodies targeting apolipoprotein A-1 (anti-apoA-1 IgGs), the major protein constituent of the high-density lipoprotein (HDL), present a growing interest. Indeed, in the last decade, numerous clinical studies performed on different CVD risk populations revealed that high levels of anti-apoA-1 IgGs were an independent CV risk factor predicting poorer CV outcomes in the general population and in high-risk populations [11-15]. In HIV, the prevalence of high anti-apoA-1 IgG levels was found to be elevated (58 %) and independently associated with high viremia load, high pro-inflammatory cytokine profile and low CD4+ cell count, three known risk markers for CV events in HIV patients [16].
Translational studies pointed to anti-apoA-1 IgGs as causal mediators of inflammation and atherogenesis [17-20]. These antibodies target innate immune receptors on macrophages inducing a high pro-inflammatory cytokine response and elicit their transformation in foam cells, two macrophage-related mechanisms that accelerate atherosclerosis plaque progression [21]. Some of these pathologic effects can be specifically blocked by a 26 amino-acid peptide derived from the C-terminal alpha loop of apoA-1(cterA1 peptide), known to be preferentially recognized by human anti-apoA-1 antibodies [22,23]. CterA1 peptide was also able to inhibit the fixation of these anti-apoA-1 IgGs to apoA-1 in in vitro ELISA assays [22]. Thereby, cterA1 peptides present a high potential to be used for both the detection of anti-apoA-1 IgGs and the neutralization of their pathogenic effects.
Therefore, in this study we investigated the prevalence of anti- C terminal apoA-1 (cterA1) IgGs in HIV patients using cterA1 peptide as a capture antigen and explored their possible associations with biological markers of HIV infection and atherosclerosis development. We also assessed the therapeutic potential of our cterA1 mimetic peptide to prevent foam cell formation induced by anti-apoA-1 IgGs and HIV-RNA in vitro [23].
Patients population and study design
This study is an ancillary study derived from [16]. Briefly, blood samples from HIV-infected individuals included in the Swiss HIV Cohort Study (http://www.shcs.ch) were collected in 2014 at Geneva University hospitals. All patients gave written informed consent for the study. The study protocol was approved by the ethics committee of Geneva University Hospitals. Exclusion criteria were: age < 40 years old, preexisting CV disease or use of lipid-lowering therapy.
Whole blood and EDTA plasma were collected and centrifuged at 800g for 10 min within 6 h of collection for measuring anti- cterA1 IgGs, fasting total cholesterol (TC), HDL, triglycerides, glucose, CD4 lymphocyte count and HIV RNA copies. LDL cholesterol levels were calculated using the Friedwald formula.
Biomarker measurements
Anti-cterA1 IgG levels were quantified in HIV patient plasma by ELISA using cterA1 peptide (20 µg/ml) as coating target. The cterA1 peptide synthesis and ELISA procedure were previously described in [22]. Anti-cterA1 IgGs were expressed as arbitrary units (OD405 nm). The positive cut-off for Anti-cterA1 IgGs was set at an OD value of 0.5, corresponding of the 97.5th percentile of the distribution obtained on healthy blood donors [24]. The specificity of our ELISA to detect antibodies against cterA1 peptide, has been demonstrated previously by LC-MS analyses and competition assays [22,23,25]. The intra and inter-assay coefficients of variation (CV) at the seropositivity cut-off were <6%.
The cytokines (IL-8, IL-10, TNF-α, MCP-1 and CRP) were quantified using the Meso Scale Discovery (MSD) platform (Rockville, MD, USA). Analyte concentrations were determined with Discovery Workbench® software 4.0, using a 4-parameter logistic fit model. The lower limit of detection in pg/ml was: IL-8, 0.04; IL-10, 0.03; TNF-α 0.04; CRP, 1.33; MCP-1, 0.09. Intra-run CVs were below 7% and inter-run CVs below 15%.
Human monocyte-derived macrophage (HMDM) preparation
Human monocytes were isolated from buffy coats obtained from healthy donors at the Geneva Hospital Blood Transfusion Center (Switzerland) as previously described [21], and differentiated into macrophages by incubation with 50 ng/ml of human macrophage colony stimulating factor (M-CSF) (PeproTech, Rocky Hill, NJ) in complete RPMI-1640-Glutamax I culture medium (10% heat-inactivated FBS, 50 µg/ml streptomycin, 50 U/ml penicillin) for 6 days.
HMDM stimulation and lipid droplet quantification
After purification, monocytes were added at 5.104 cell/well to 8 well Lab-Tek™ II chamber slide (Thermo scientific, Nunc™) in complete medium + M-CSF until macrophage differentiation.
For these in vitro studies, commercial polyclonal goat anti-human apoA-1 IgGs were used as surrogates for human anti-apoA-1 IgGs as they were thoroughly validated to have the same specificity and the same pathological activity in vivo and in vitro as anti-apoA-1 IgGs isolated from patients [17,21-23]. These polyclonal goat anti-human apoA-1 IgGs recognize cterA1 peptides in ELISA test [22].
HMDM were treated with 1 µg/ml of ssRNA40/LyoVec™ (ssRNA40 is a single stranded GU-rich oligonucleotide derived from the HIV long terminal repeat that mimics HIV cell stimulation via TLR7/8) or control ssRNA41/LyoVec™ (Invivo Gen, San Diego, CA) [24], with 50 µg/ml of polyclonal goat anti-apoA-1 IgGs (Academy Bio-Medical Company, Inc., Houston, TX) or polyclonal goat control IgGs (CTL IgG) (Meridian Life Science, Saco, ME) for 24 h. In inhibition experiments, cterA1 peptide or control scramble peptide (peptide corresponding to a random amino-acid sequence of cterA1 peptide) were incubated at a concentration of 100 µg/ml together with anti-apoA-1 IgG or CTL IgG, or with ssRNA40 or control ssRNA41 at concentration of 1 ug/ml, during 1 h prior addition of the mix to the HMDM [22].
At the end of the incubation period, HMDM were washed 3 times with PBS and fixed with 4%PAF for 10 min.
To identify the formation of foam cell, lipids stocked in cytosolic droplets were labelled with BODIPY™ 493/503 dye (ThermoFisher Scientific), that specifically stained triglycerides and cholesterol esters [26], at 1 ug/ml for 30 min. After three PBS washes, the slides were mounted with VectaShield® antifade mounting medium + DAPI (Vector Laboratories, Inc. Burlingame, CA). Images were taken with a LSM700 Meta Confocal microscope (Carl Zeiss AG, Germany) using x60 objective (EC Plan Neofluar x40 1.3 Oil DIC, Zeiss). The quantification of the fluorescent lipid droplets per cell, identified by DAPI blue nuclear staining, was performed using the MetaMorph® v7.7 software (Visitron Systems GmbH, Germany). The results were expressed as the mean fluorescence intensity (MFI) calculated using the fluorescent surface area of the droplets per cell on each picture taken and for each condition. 10 pictures were taken per condition for each experiment and a total of 3 to 7 independent experiments were performed. Examples of images were resumed in supplementary Figures 1, 2 and 3.
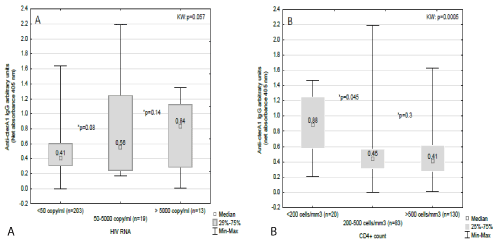
Figure 1. Evolution of anti-cterA1 IgG according to HIV verimia strata (a) and CD4+ count (b)
Data are presented in Box and Whisker graph with median, IDR and range. *p values according to Mann-Whitney test. KW: Kruskall-Wallis test.
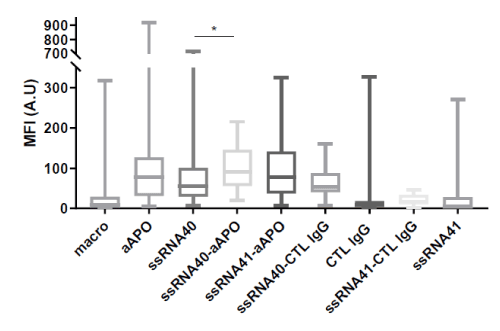
Figure 2. Lipid droplet formation in human macrophages induced by ssRNA40 in presence or not of anti-apoA-1 IgG
ssRNA40 or its control ssRNA41 (1µg/ml) and polyclonal goat anti-apoA-1 IgGs (aAPO) or polyclonal goat CTL IgGs (50 µg/ml) were added individually or in combination, as indicated in the graph, on macrophages for 24 h. Lipid-loaded droplets were stained with BODIPY 493/503 dye. The results are expressed as the mean fluorescence intensity calculated as described in Materials and Methods. Data are presented in Box and Whisker graph with median, IQR and range. Significant difference between conditions was assessed by Mann-Whitney test on 3 to 8 independent experiments. All the conditions with aAPO and ssRNA40 are significantly different from the controls with p<0.0001. The significance for difference between ssRNA40 and ssRNA40-aAPO conditions is *p=0.0192.
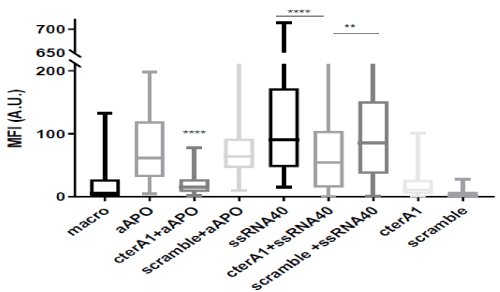
Figure 3. C-terA1 peptides inhibited lipid droplet formation in macrophages
Macrophages were treated 24 h with (A) anti-apoA-1 IgGs (50 µg/ml) preincubated with or without cterA1 or scramble peptides (100 µg/ml) for 1 h or with (B) ssRNA40 (1µg/ml) preincubated with or without cterA1 or scramble peptides(1 µg/ml) 1 h . Lipid droplets were stained with BODIPY 493/503 dye and fluorescence quantified as described in Materials and Methods. Data are presented in Box and Whisker graph with median, IQR and range. Significant difference between conditions was assessed by Mann-Whitney test on 7 independent experiments. (**p=0.0050;****p<0.0001).
Statistics
Exact bilateral Fischer test and Mann–Whitney test were used when appropriate to compare the different groups of patients. Spearman rank correlations were performed to establish correlation between variables. Because of the exploratory nature of this study and chosen variables investigated, correction for multiple testing was not performed. Kruskall-Wallis test was used to detect a trend in the dose-response experiments. Logistic regression was used to determine the risk of having CD4+ counts < 200 cells/mm3 and a detectable viremia according to anti-apoA-1 IgG status. Results were expressed as adjusted odds ratios (OR), with 95% confidence intervals (95% CI) adjusting for viremia, ART and inflammation cytokine profile. Sensitivity analyses were not performed because of the limited sample size of this pilot study. A 2-sided p-value <0.05 was considered statistically significant. Analyses were performed with Statistics (version 8.0). For in vitro studies, statistical analyses were performed using Mann-Whitney test with Prism 6.
Characteristic of HIV patients according to anti-cterA1 IgG status
High anti-cterA1 IgG levels were detected in 62 out of the 237 HIV patients, representing 26% (95%CI: 21-32%) of anti- cterA1 IgG seropositivity, as indicated in Table 1. There were no significant differences between anti-cterA1 IgG positive and anti-cterA1 IgG negative patients in terms of baseline demographic characteristics, ART treatment, traditional CV risk factors, and 10-years Framingham risk score (FRS) or dyslipidaemia (Table 1). However, patients positive for anti-cterA1 antibodies presented a significantly lower CD4+ cell count, a higher number of detectable HIV-RNA copies, and a significant increase of IL-8 and TNF-α cytokines (Table 2). Furthermore, spearman correlations showed significant association between anti-cterA1 IgG antibodies levels and the number of HIV RNA copies, CD4+ count and the level of IL-8 and TNF-α cytokines (Table 3).
Table 1. Demographic characteristics according to anti-cterA1 IgG status
|
HIV
(n=237) |
HIV patients anti-cterA1 + (n=62) |
HIV patients anti-cterA1 - (n=175) |
P |
Age |
49 (45-53) |
49 (45-55) |
49 (44-53) |
0.64 |
Male Gender, % (n) |
66.6 (158) |
69 (43) |
65 (115) |
0.64 |
Weight, kg |
74 (65-83) |
71 (64-80) |
74 (65-85) |
0.25 |
Waist, cm |
90 (83-100) |
90 (84-99) |
90 (83-100) |
0.73 |
Hip, cm |
98 (92-105) |
95 (90-102) |
99.5 (92-105.5) |
0.075 |
BMI, kg/m2 |
25 (22.5-28.3) |
24.6 (21.8-27.8) |
25.2 (22.8-28.4) |
0.29 |
Systolic BP, mmHg |
127 (117-135) |
127.5 (116.5-131) |
126.5 (117-136.5) |
0.46 |
Diastolic BP, mmHg |
80 (73-86) |
79 (73-86) |
80 (74-86) |
0.68 |
Smoke, % (n) |
35.8 (85) |
40.3 (25) |
34.2 (60) |
0.44 |
Fat loss, % (n) |
14.3 (34) |
12.9 (8) |
14.85 (26) |
0.83 |
Fat accumulation, % (n) |
30.8 (73) |
25.8 (16) |
32.5 (57) |
0.34 |
Depression, % (n) |
17.2 (41) |
14.5 (9) |
18.3 (32) |
0.56 |
Familial history of CAD*, % (n) |
8.4 (20) |
12.9 (8) |
6.8 (12) |
0.29 |
Framingham risk score, % |
8.4 (5-17) |
7 (1.56-15.52) |
8.2 (1.38-14.2) |
0.8 |
Treatment |
NRTI, % (n)† |
90 (200) |
79 (49) |
86(151) |
0.22 |
NNRTI, % (n) |
49.3 (117) |
37 (23) |
53.7(94) |
0.76 |
PI, % (n) |
28.2 (67) |
25.8 (16) |
29 (51) |
0.74 |
Integrase, % (n)† |
15.9 (35) |
15(8) ‡ |
16 (27)§ |
1.0 |
Auto-antibodies |
Anti-cterA1 IgG, OD (range) |
0.98 (0.64-2.19) |
- |
- |
- |
Anti-cterA1 IgG positivity % (n) |
26 (62) |
- |
- |
- |
*undetermined in 9 patients; Data are expressed as median and (interquartile range) unless stated otherwise
†undetermined in 17 patients; ‡ undetermined in 9 patients; § undetermined in 8 patients
Exact two-tailed fisher for categorical variable- MW for continuous variables |
Table 2. Biological characteristics according to anti-cterA1 IgG status (new cut-off)
|
HIV (n=237) |
HIV patients anti-cterA1 + (n=62) |
HIV patients anti-cterA1 - (n=175) |
P |
Hematology |
Hb, g/L |
14.5 (13-15.3) |
13.9 (12.7-15.2) |
14.2 (13.1-15.3) |
0.24 |
Leuco, G/L |
5800 (4600-6900) |
5350 (4400-7200) |
5900 (4700-6900) |
0.44 |
Platelets, G/L |
223.5 (186-268) |
211.5 (182-245) |
229 (191-278) |
0.053 |
Lympho G/L |
1833 (1476-2439) |
1692 (1372-2347) |
1893 (1528.5-2458.5) |
0.113 |
Lympho % |
35.9 (29.3-41.9) |
34.7 (29.8-42) |
36.4 (28.6-41.8) |
0.65 |
CD4 cells/mm3 |
533 (351.5-761.5) |
475 (236-687) |
546.5 (401.5-778) |
0.0132 |
CD4% |
30 (22.5-36) |
26 (15-35) |
30 (25-37) |
0.0052 |
Virology |
Detectable HIV RNA, (copy/ml ) |
50594 (180000) |
98982 (250000) |
7899.5 (18372) |
0.057 |
HIV RNA copy <50/ml; % (n) |
85.6 (203) |
74.2 (46) |
89.7 (157) |
<0.0053 |
Lipid profile |
Cholesterol, mmol/l |
5.0 (4.2-5.7) |
4.65 (4-5.4) |
5.15 (4.35-6) |
0.0041 |
HDL, mmol/l |
1.19 (1.0-1.48) |
1.195 (0.97-1.65) |
1.19 (1-1.44) |
0.64 |
Triglycerids, mmol/l |
1.32 (0.94-2.05) |
1.22 (0.84-1.78) |
1.36 (0.96-2.1) |
0.074 |
LDL, mmol/l |
3.0 (2.36-3.7) |
2.76 (2.23-3.22) |
3.04 (2.47-3.8) |
0.027 |
PCSK-9, ng/ml |
256 (214-323) |
273 (225-313.6) |
256.3 (214.4-323.5) |
0.89 |
Liver function |
ASAT, U/l |
21 (15-27) |
23 (18-31) |
20 (15-27) |
0.038 |
ALAT, U/l |
24 (17-35) |
24 (15-33.5) |
24 (17-35) |
0.96 |
Renal function |
Creatinine, µmol/l |
76 (65-88) |
74 (64-87) |
76.5 (65-88) |
0.46 |
Inflammation |
IL-8, pg/ml |
2.45 (1.3-4.88) |
3.35 (1.83-6.68) |
2.17 (1.19-4.18) |
0.0012 |
IL-10, pg/ml |
0.16 (0.11-0.25) |
0.186 (0.13-0.43) |
0.15 (0.10-0.21) |
0.0079 |
TNF-a, pg/ml |
1.71 (1.3-2.3) |
2.27 (1.6-3) |
1.63 (1.29-1.98) |
<0.0001 |
MCP-1, pg/ml |
128.1 (95-166.8) |
129.8 (92.7-166.7) |
126.2 (95-167) |
0.97 |
CRP, mg/ml |
2.42 (0.83-6.64) |
3.76 (1.13-6.64) |
2.0 (0.81-6.83) |
0.158 |
Data are expressed as median and (interquartile range) unless stated otherwise |
Table 3. Spearman correlations between anti-cterA1 IgG and conventional biological parameters
|
Spearman correlations |
Anti-cterA1 IgG, OD |
r; p value |
Hb, g/L |
-0.075; p=0.25 |
Leucocytes, G/L |
-0.05; p=0.44 |
Platelet, G/L |
-0.12; p=0.053 |
Lymphocytes, G/L |
-0.099; p=0.13 |
Lymphocytes, % |
-0.03; p=0.65 |
CD4, cells/mm3 |
-0.16; p=0.013 |
CD4, % |
-0.182; p=0.005 |
HIV RNA, copy /ml |
0.198; p=0.002 |
Cholesterol, mmol/l |
-0.19; p=0.004 |
HDL, mmol/l |
0.024; p=0.72 |
LDL, mmol/l |
-0.15; p=0.027 |
Triglycerids |
-0.12; p=0.074 |
ASAT, U/L |
0.138; p=0.03 |
ALAT, U/l |
-0.007; p=0.91 |
Creatinine µmol/l |
-0.05; p=0.43 |
PCSK-9, ng/ml |
0.009; p=0.89 |
FRS |
-0.016 ; p=0.8 |
IL-8, pg/ml |
0.2; p=0.0011 |
IL-10, pg/ml |
0.17; p=0.007 |
TNF-a, pg/ml |
0.29; p<0.0001 |
MCP-1, pg/ml |
0.002; p=0.975 |
CRP, mg/ml |
0.09; p=0.158 |
Anti-cterA1antibody evolution across HIV viremia and CD4+ lymphocyte strata
We looked at the distribution of anti-cterA1 IgGs according to HIV-RNA copy strata (< 50; 50-5000; >5000 copies/ml) and CD4+ lymphocyte strata (<200; 200-500; >500 cells/mm3). As show in Figure 1, the level of anti-cterA1 IgGs increased concomitantly with the HIV replication status (Figure 1A) and inversely, anti-cterA1 IgG titre decreased proportionally to the increasing CD4+ lymphocytes count (Figure 1B).
Anti-cterA1 IgG as predictors of HIV viremia and CD4+ lymphopenia
We performed logistic regression analysis to determine whether anti-cterA1 IgGs could be predictors of high HIV viremia and low CD4+ count. Anti-cterA1 IgGs were associated with a 6-fold increased risk of having CD4+ count <200 cells/mm3 (OR: 6.38; 95%CI: 2.41-16.90; p=0.0001), which remained similar after adjustment for HIV viremia and current HAART (adjusted OR: 5.02;95%CI:1.73-14.54; p=0.0029), and with a 3-fold increased risk of high viremia load (OR: 3.08; 95%CI:1.45-6.52; p=0.003), which stayed of the same order of magnitude after adjustment for HAART (adjusted OR: 2.57; 95%CI: 1.10-6.03; p=0.029).
CterA1 peptide as a potential inhibitor of foam cell formation induced by anti-apoA-1 IgG and ssRNA40 in vitro
As HIV patients positive for anti-cterA1 (and anti-apoA-1) IgGs were found to have specific HIV biological characteristics known to be associated with increased cardiovascular risk (higher viremia, lower CD4+ lymphocytes count, higher levels of pro-inflammatory cytokines [4-6], and because previous studies shown that cterA1 peptide reversed the proinflammatory effects of anti-apoA-1 IgGs on macrophages [22], we investigated whether anti-apoA-1 IgGs (polyclonal goat IgG from commercial source) and HIV single strand RNA (ssRNA) 40 could influence the formation of foam cells, a hallmark of atherosclerosis, in a cterA1 peptide-dependent manner. In this perspective, HMDM were incubated with ssRNA40 or polyclonal goat anti-apoA-1 IgG alone or with a mix of them (and their control counterpart: polyclonal goat CTL IgG and scramble ssRNA41) for 24 h at optimal concentrations defined previously [21,22,24]. The increased uptake of lipids following macrophage stimulation results in the formation of cytosolic droplets characterizing foam cells, that were labelled with the lipophilic fluorescent dye BODIPY 493/503 (see supplementary Figures 1, 2 and 3 for illustration).
As shown in Figure 2, incubation of HMDM with either polyclonal goat anti-apoA-1 IgGs or ssRNA40 significantly increased lipid droplet accumulation in a similar manner, showing the transformation of macrophages to foam cells. The concomitant addition of polyclonal goat anti-apoA-1 IgGs and ssRNA40 resulted in a non-significant increase of lipid droplet formation. None of the controls (ssRNA41 or CTL IgG) had any effect of HMDM lipid accumulation.
CterA1 peptide or its control counterpart (scramble peptide) were then co-incubated 1 h with the polyclonal goat anti-apoA-1 IgGs or ssRNA40 prior their addition to the HMDM. As shown in Figure 3, cterA1 peptide co-incubation reduced the anti-apoA-1 IgGs and ssRN40-induced lipid droplet formation, whereas no difference was found when anti-apoA-1 IgGs and ssRNA40 were co-incubated with the scramble peptide.
The first notable finding of this study is that the prevalence of high anti-cterA1 IgG levels (seropositivity) is substantial in HIV patients, reaching 26%. Such prevalence is similar to what has recently been reported in patients suffering from systemic lupus erythematosus (22%), where these antibodies were found to be associated with disease severity [25]. Of note, the prevalence of anti-cterA1 IgG levels in HIV is roughly half of what has been retrieved for anti-apoA-1 IgG on the same cohort (58%) [16], which can be explained by the fact that by using a cterA1 peptide as a coating antigen, we do not capture the totality of the polyclonal anti-apoA-1 IgG repertoire. Nevertheless, we were able to reproduce the marked associations between anti-cterA1 IgG seropositivity and higher viremia and lower CD4+ count, which were independent of HAART and viremia respectively, as previously shown for anti-apoA-1 IgG [16]. Furthermore, the associations between anti-cterA1 IgG and several markers of inflammation (TNF-α, IL-8 and IL-10) were also replicated and of the same order of magnitude than what has been reported previously [16,20,21]. On the other hand, the inverse associations between anti-cterA1 IgG with total and LDL cholesterol in HIV have not been reported with anti-apoA-1 IgG before [16], similarly to what has been retrieved in a case-control study where anti-cterA1 IgG, but not anti-apoA-1 IgG, were found to be inversely associated with lipids fractions associated to apoB particles [27]. Whether anti-cterA1 IgG represent or not a subgroup of anti-apoA-1 antibodies associated with specific lipid metabolism abnormalities or disease is still elusive and warrants further investigations. Taken together, these results reinforce the hypothesis that cterA1 mimetic peptide in a given three-dimensional conformation could be used in vitro assay to detect anti-apoA-1 IgG [22], despite displaying a lower seropositivity prevalence (26% vs. 58%).
The second important finding of this study resides in the observation that our cterA1 mimetic peptide could reverse the HIV ssRNA and anti-apoA-1 IgG-induced foam cell transformation in vitro, whereas the corresponding scramble peptide did not. Aside the fact that macrophage foam cell formation is the hall-mark of atherogenesis [28], the possible relevance of this observation is further reinforced by the fact that both HIV-ssRNA and anti-apoA-1 IgG have been proposed to play a causal role in atherogenesis [17,29]. On one hand, HIV-ssRNA was shown to induce pro-inflammation cytokine production and lipid up-loading in macrophages through a Toll-like receptor (TLR) 8-dependent activation, in in vitro studies [24], which may at least partly explain the increased CV risk in HIV individuals [4,6,30]. On the other hand, the direct pro-atherogenic role of anti-apoA-1 IgG has been shown to be mediated by TLR2/TLR4/CD14 complex, leading to inflammation, increased foam cell formation in vitro, and increased propensity to atherogenesis, myocardial necrosis and death in mice [17-19,21]. These mechanistic studies provided a molecular rationale for the poor and independent CV prognosis ascribed to these autoantibodies in humans [11-14].
Because cterA1 peptide was shown to inhibit the production of various pro-atherogenic cytokines [22], we currently believe that cterA1 peptides reverse foam cell formation by a cytokine-mediated pro-atherogenic effect, but we cannot exclude the fact that cterA1 peptide may act on macrophage lipid metabolism, which is currently under investigations. Along the same line, the molecular mechanisms by which cterA1 peptide can reverse the HIV-induced foam cell formation are currently elusive. We might hypothesize that, because of its alpha-helix structure with high content of aliphatic components [22], the peptide has a high potential to perturb membrane integrity and interfere with membrane-particle fusion, which may be the case for the HIV-ssRNA40 as it is integrated in a lipid vehicle for better capture by the cells, but this assumption is currently devoid of any experimental evidence.
Finally, because anti-cterA1 IgG positively correlated with HIV RNA load in HIV patients, we investigated whether the concomitant presence of both agents may exacerbate foam cell formation, but we did not observe any cumulative or synergistic effects when anti-cterA1 IgG and ssRNA40 were simultaneously added on human macrophages. As both ssRN40 and anti-apoA-1 IgGs are known to activate Toll-like receptors, this non-cumulative effect observed might be explained in part by the cross-tolerance mechanism described for TLRs, which implies that once a TLR is activated, the triggered Myd88-dependent intracellular pathway, common to most of TLRs, is refractory to subsequent activation from other TLRs, which is the case for TLR2/TLR4 and TLR8 [31]. Extrapolated to HIV patients, one may consider that the absence of synergistic effects between ssRNA40 and anti-apoA-1 IgG on foam cell formation would not make a strong case for anti-apoA-1/cterA1 seropositive individuals to be at higher CV risk than anti-apoA-1/cterA1 seronegative patients.
Nevertheless, despite these important remaining questions, our results indicate that the cterA1 peptide ability to reverse foam cell formation in vitro has the potential to open new therapeutic perspectives in the field of CVD in HIV by targeting both the pro-atherogenic effects of anti-apoA-1/ cterA1 IgG and HIV RNA.
This study presents various limitations. Firstly, due to the cross-sectional nature of our study which included relatively young HIV individuals, the absence of follow-up or radiological assessment of atherosclerosis burden, we could not relate the presence of HIV-RNA load or anti-cterA1 levels to anamnestic, incident CV events or to the presence of asymptomatic atherosclerosis. Therefore, the demonstration of the added overall prognostic value of measuring anti-A-1/cterA1 IgG in HIV individuals remains to be demonstrated in the first place. Regarding the in vitro experiments, we used an artificial relipidated HIV-derived ssRNA as TLR8 agonist instead of a complete viral particle. As so, this model took abstraction of the implication of HIV proteins, especially Nef and Tat, in the modulation of macrophage intracellular signalling, which should have certainly influence the overall inflammatory response of macrophages with a probable echo on lipid uptake [24,32], and gp120/41 for the management of HIV particle fusion to macrophage membrane. Although, apoA-1 whole molecule has been reported to inhibit HIV particle fusion with host cell and syncytium formation by different mechanisms involving direct binding to gp120/41 with subsequent blockage of the protein interaction with its host cell receptor or binding to the lipid membrane surface and stopping fusion process [33-35], a more in deep study should be performed to confirm the inhibitory function of cterA1 peptide of HIV particle fusion.
In conclusion, high levels of anti-cterA1 IgGs are associated with biological HIV disease severity and with a pro-inflammatory biological profile. By reversing the HIV ssRNA40-and anti-apoA-1 IgG-induced foam cell formation, our cterA1 mimetic peptide has the potential to represent a specific anti-atherosclerotic therapeutic agent in HIV settings.
NV and SP are named as co-inventors of the patent related to cterA1 peptide (“Mimetic peptides for prognosis, diagnosis or treatment of a cardiovascular disease”, N° P1347EP00).
Authors have no conflict of interest to declare.
We thank all participants, doctors and nurses from the Five Swiss University Hospitals, two Cantonal Hospitals, 15 affiliated hospitals and 36 private physicians that are associated with the Swiss HIV Cohort Study (SHCS). The members of the Swiss HIV Cohort Study are: Aubert V, Barth J, Battegay M, Bernasconi E, Böni J, Bucher HC, Burton-Jeangros C, Calmy A, Cavassini M, Egger M, Elzi L, Fehr J, Fellay J, Francioli P, Furrer H (Chairman of the Clinical and Laboratory Committee), Fux CA, Gorgievski M, Günthard H (President of the SHCS), Haerry D (deputy of "Positive Council"), Hasse B, Hirsch HH, Hirschel B, Hösli I, Kahlert C, Kaiser L, Keiser O, Kind C, Klimkait T, Kovari H, Ledergerber B, Martinetti G, Martinez de Tejada B, Metzner K, Müller N, Nadal D, Pantaleo G, Rauch A (Chairman of the Scientific Board), Regenass S, Rickenbach M (Head of Data Center), Rudin C (Chairman of the Mother & Child Substudy), Schmid P, Schultze D, Schöni-Affolter F, Schüpbach J, Speck R, Taffé P, Tarr P, Telenti A, Trkola A, Vernazza P, Weber R, Yerly S.
This work was supported by European Commission (FP7-INNOVATION I HEALTH-F2-2013-602114; Athero-B-Cell: Targeting and exploiting B cell function for treatment in cardiovascular disease), by Swiss National Science Foundation Grants [#310030_152912/1 to F.M. ; #310030_163335 to N.V.], by a grant from the Leenaards Foundation [#3698 to N.V.], by the Swiss HIV Cohort Study financed by the Swiss National Science Foundation [grant #148522] and by the SHCS Research Foundation.
N.S. and S.P. performed the experiments, data analyses, interpretation, and manuscript redaction. N.V. and A.C. contributed to study design, data analyses and manuscript redaction. A.C., L.K. and B.G. provided cohort samples. O.H. provided the cterA1 peptide. F.M, L.K. and B.G performed critical revising of the manuscript.
- Hemkens LG, Bucher HC (2014) HIV infection and cardiovascular disease. Eur Heart J 35: 1373-1381. [Crossref]
- Boccara F, Cohen A (2016) HIV and Heart Disease: What Cardiologists Should Know. Rev Esp Cardiol 69: 1126-1130. [Crossref]
- Jain A, Kolvekar T, Nair DR (2018) HIV infection and lipids. Curr Opin Cardiol 33: 429-435. [Crossref]
- Calmy A, Gayet-Ageron A, Montecucco F, Nguyen A, Mach F, et al. (2009) HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 23: 929-939. [Crossref]
- Augustemark De Lima LR, Petroski EL, Moreno YMF, Silva DAS, Trindade EBMS, et al. (2018) Dyslipidemia, chronic inflammation, and subclinical atherosclerosis in children and adolescents infected with HIV: The Positive Health Study. PLoS One 13: e0190785. [Crossref]
- Hsu DC, Ma YF, Hur S, Li D, Rupert A, et al. (2016) Plasma IL-6 levels are independently associated with atherosclerosis and mortality in HIV-infected individuals on suppressive antiretroviral therapy. AIDS 30: 2065-2074. [Crossref]
- Da Cunha J, Ferreira Maselli LM, Treitinger A, Monteiro AM, Gidlund M, et al. (2013) Serum levels of IgG antibodies against oxidized LDL and atherogenic indices in HIV-1-infected patients treated with protease inhibitors. Clin Chem Lab Med 51: 371-378. [Crossref]
- Füst G, Beck Z, Bánhegyi D, Kocsis J, Bíró A, et al. (2005) Antibodies against heat shock proteins and cholesterol in HIV infection. Mol Immunol 42: 79-85. [Crossref]
- Orellana RV, Fonseca HA, Monteiro AM, Ortega KL, Gallottini MH, et al. (2013) Association of autoantibodies anti-OxLDL and markers of inflammation with stage of HIV infection. Int J Cardiol 168: 1610-1612. [Crossref]
- Vega LE, Espinoza LR (2018) HIV infection and its effects on the development of autoimmune disorders. Pharmacol Res 129: 1-9. [Crossref]
- Vuilleumier N, Bas S, Pagano S, Montecucco F, Guerne PA, et al. (2010) Anti-apolipoprotein A-1 IgG predicts major cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum 62: 2640-2650. [Crossref]
- Vuilleumier N, Rossier MF, Pagano S, Python M, Charbonney E, et al. (2010) Anti-apolipoprotein A-1 IgG as an independent cardiovascular prognostic marker affecting basal heart rate in myocardial infarction. Eur Heart J 31: 815-823. [Crossref]
- Vuilleumier N, Pagano S, Combescure C, Gencer B, Virzi J, et al. (2019) Non-Linear Relationship between Anti-Apolipoprotein A-1 IgGs and Cardiovascular Outcomes in Patients with Acute Coronary Syndromes. J Clin Med 8: E1002. [Crossref]
- Anderson JLC, Pagano S, Virzi J, Dullaart RPF, Annema W, et al. (2019) Autoantibodies to Apolipoprotein A-1 as Independent Predictors of Cardiovascular Mortality in Renal Transplant Recipients. J Clin Med 8: E948. [Crossref]
- Antiochos P, Marques-Vidal P, Virzi J, Pagano S, Satta N, et al. (2017) Impact of CD14 Polymorphisms on Anti-Apo A-1 (Apolipoprotein A-1) IgG-Related Coronary Artery Disease Prediction in the General Population. Arterioscler Thromb Vasc Biol 37: 2342-2349. [Crossref]
- Satta N, Pagano S, Montecucco F, Gencer B, Mach F, et al. (2018) Anti-apolipoprotein A-1 autoantibodies are associated with immunodeficiency and systemic inflammation in HIV patients. J Infect 76: 186-195. [Crossref]
- Pagano S, Carbone F, Burger F, Roth A, Bertolotto M, et al. (2016) Anti-apolipoprotein A-1 auto-antibodies as active modulators of atherothrombosis. Thromb Haemost 116: 554-564. [Crossref]
- Montecucco F, Braunersreuther V, Burger F, Lenglet S, Pelli G, et al. (2015) Anti-apoA-1 auto-antibodies increase mouse atherosclerotic plaque vulnerability, myocardial necrosis and mortality triggering TLR2 and TLR4. Thromb Haemost 114: 410-422. [Crossref]
- Montecucco F, Vuilleumier N, Pagano S, Lenglet S, Bertolotto M, et al. (2011) Anti-Apolipoprotein A-1 auto-antibodies are active mediators of atherosclerotic plaque vulnerability. Eur Heart J 32: 412-421. [Crossref]
- Vuilleumier N, Pagano S, Montecucco F, Quercioli A, Schindler TH, et al. (2019)b Relationship between HDL Cholesterol Efflux Capacity, Calcium Coronary Artery Content, and Antibodies against ApolipoproteinA-1 in Obese and Healthy Subjects. J Clin Med 8: E1225. [Crossref]
- Pagano S, Satta N, Werling D, Offord V, De Moerloose P, et al. (2012) Anti-apolipoprotein A-1 IgG in patients with myocardial infarction promotes inflammation through TLR2/CD14 complex. J Intern Med 272: 344-357. [Crossref]
- Pagano S, Gaertner H, Cerini F, Mannic T, Satta N, et al. (2015) The Human Autoantibody Response to Apolipoprotein A-I Is Focused on the C-Terminal Helix: A New Rationale for Diagnosis and Treatment of Cardiovascular Disease?. PLoS One 10: e0132780. [Crossref]
- Teixeira PC, Ducret A, Ferber P, Gaertner H, Hartley O, et al. (2014) Definition of human apolipoprotein A-I epitopes recognized by autoantibodies present in patients with cardiovascular diseases. J Biol Chem 289: 28249-28259. [Crossref]
- Bernard MA, Han X, Inderbitzin S, Agbim I, Zhao H, et al. (2014) HIV-derived ssRNA binds to TLR8 to induce inflammation-driven macrophage foam cell formation. PLoS One 9: e104039. [Crossref]
- Nigolian H, Ribi C, Courvoisier DS, Pagano S, Alvarez M, et al. (2019) Anti-apolipoprotein A-1 autoantibodies correlate with disease activity in systemic lupus erythematosus. Rheumatology pii: kez306. [Crossref]
- Qiu B, Simon MC (2016) BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio Protoc 6: e1912. [Crossref]
- Dullaart RPF, Pagano S, Perton FG, Vuilleumier N (2019) Antibodies Against the C-Terminus of ApoA-1 Are Inversely Associated with Cholesterol Efflux Capacity and HDL Metabolism in Subjects with and without Type 2 Diabetes Mellitus. Int J Mol Sci 20: E732. [Crossref]
- Hansson GK, Hermansson A (2011) The immune system in atherosclerosis. Nat Immunol 12: 204-212. [Crossref]
- Oliviero U, Bonadies G, Apuzzi V, Foggia M, Bosso G, et al. (2009) Human immunodeficiency virus per se exerts atherogenic effects. Atherosclerosis 204: 586-589. [Crossref] 29
- Alcaide ML, Rodriguez VJ, Abbamonte JM, Pallikuth S, Langlie J, et al. (2019) HIV and carotid atherosclerosis: a mediational model. AIDS Care 23: 1-5. [Crossref] 30
- Akira S, Sato S (2003) Toll-like receptors and their signaling mechanisms. Scand J Infect Dis 35: 555-562. [Crossref]
- Herbein G, Gras G, KHAN KA, Abbas W (2010) Macrophage signaling in HIV-1 infection. Retrovirology 7: 34. [Crossref]
- Martin I, Dubois MC, Saermark T, Ruysschaert JM (1992) Apolipoprotein A-1 interacts with the N-terminal fusogenic domains of SIV (simian immunodeficiency virus) GP32 and HIV (human immunodeficiency virus) GP41: implications in viral entry. Biochem Biophys Res Commun 186: 95-101. [Crossref]
- Owens BJ, Anantharamaiah GM, Kahlon JB, Srinivas RV, Compans RW, et al. (1990) Apolipoprotein A-I and its amphipathic helix peptide analogues inhibit human immunodeficiency virus-induced syncytium formation. J Clin Invest 86: 1142-1150. [Crossref]
- Panin LE, Kostina NE, Lukashev VA (2002) The role of structural and functional homology between human apolipoprotein A-I and envelope proteins of human immunodeficiency virus type 1 in CD4 receptor binding. Dokl Biochem Biophys 385: 209-212. [Crossref]