Hemophagocytic syndrome is a rare, multifactorial entity characterized by an exaggerated activation of macrophages, which leads to an excessive release of cytokines mediated by T lymphocytes, with high mortality, with a mean survival without treatment of less than 2 months. We present the case of a young woman with a diagnosis of pre-B lymphoblastic leukemia with clinical criteria of hemophagocytic syndrome, who died after 2 months of septic shock.
Hemophagocytic syndrome, also called hemophagocytic lymphohistiocytosis, is a rare, multifactorial pathogenetic disease characterized by an exaggerated activation of macrophages, which leads to an excessive release of cytokines mediated by T lymphocytes [1], with high mortality, presenting a mean treatment-free survival of less than 2 months [2].
Epidemiology
The incidence varies according to regions, with a predominance in pediatric patients. In adults, there has been an average age of onset at 50 years with a female predominance in a 1:7 with respect to the men [3]. An estimated worldwide incidence of 1.2 cases per million inhabitants. In Latin America, there is no standardized report that allows establishing epidemiological data, with an estimated survival of 38% one year after diagnosis [4] (Figure 1). However, this figure may be underestimated as it is a diagnosis of exclusion and due to the lack of specificity of the clinical picture.
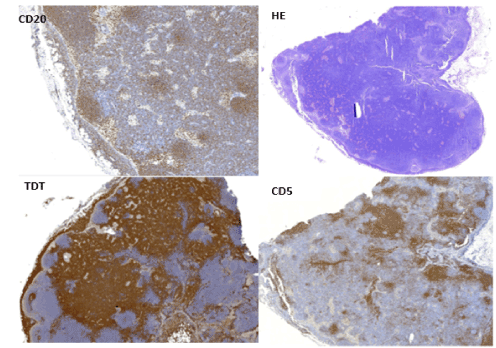
Figure 1. Immunohistochemistry with positive staining CD 20, HE, TDT, CD5
Classification
It is classified based on its causal agent as primary or secondary. The primary form (familial hemophagocytic lymphohistiocytosis) has an expression during childhood. It is caused by autosomal recessive alterations, classified into 5 types (FHL 1, FHL 2, FHL 3, FHL 4 and FHL 5) according to the associated genetic alteration that leads to an alteration of apoptosis mediated by cytotoxic T lymphocytes and Natural Killer (NK) cells. On the other hand, the secondary presentation, predominantly in adults, is associated with triggering factors such as viral (29%), bacterial, fungal, and parasitic (20%) infections; neoplasms (27%), rheumatological disorders (7%) and autoimmune syndromes (6%), as well as certain drugs, vaccines, surgery, postpartum and severe burns. Neoplasms are predominantly hematologic due to T and B cell lymphoma, or after chemotherapy treatment [5].
Physiopathology
The pathophysiological mechanism is characterized by a severe inflammatory state activated by the previously described factors, which produces a cytotoxic response mediated by CD8+ cells and NK cells, which are physiologically responsible for a lytic process through exocytosis of granules cytotoxic agents containing granzymes and perforins that enter cells to lyse them, however, there is an ineffective elimination of the antigenic stimulus by the NK cells, as well as an uncontrolled activation of the antigen-presenting cells and of the T lymphocytes activated by the stimulation Repetition of interferon gamma - dependent Toll-like receptors (IFN- γ). This sustained activation generates a humoral response with the release of pro-inflammatory cytokines (TNF-α, IFN-γ, IL-6, IL-18, and IL-12) that perpetuate antigen presentation and T-cell activation that produces a vicious cycle of inflammation and release of cytokines that generates tissue damage and multiple organ failure [6,7].
Diagnosis
The diagnosis is generally delayed due to the lack of initial suspicion due to its varied symptoms and similarity to other pathologies.
Initially, the clinical manifestations are nonspecific, resembling a serious infectious process, characterized by continuous fever (>38.5° C), lymphadenopathy and hepatosplenomegaly. In some patients, they may present with skin changes with rash, edema, purpura or petechiae. After the progression of the disease, multiple organ failure occurs with involvement of the liver, lung, and central nervous system [6].
In the initial diagnostic approach is seen in hematological parameters progressive pancytopenia according to the condition in bone marrow. The failure liver is characterized by hyperbilirubinemia, hypertransaminasemia, increased lactate dehydrogenase, hyponatremia that may be associated with inappropriate secretion of antidiuretic hormone. Likewise, there are alterations in coagulation, with hypofibrinogenemia and elevation of D-Dimer, even reaching disseminated intravascular coagulation. The hypertriglyceridemia has been associated with an inhibition of lipoprotein lipase caused by excessive TNF alpha. Pleocytosis with elevated lymphocytes, histiocytes, and proteins, as well as hemophagocytosis, can be found in cerebrospinal fluid [7].
The inflammatory response is presented with an increase in acute phase reactants (erythrocyte sedimentation rate and C-reactive protein). The hyperferritinemia, has been considered an important factor in the diagnosis, especially in high concentrations (between 7000-10000 mg/L). The immune response is determined through high concentrations of soluble CD25 (IL-2 receptor a), or soluble CD 163 (transmembrane carrier protein of hemoglobin and haptoglobin expressed on the surface of macrophages and monocytes). Low or absent NK cell activity is another diagnostic criterion [8].
The haemophagocytosis is detected in the bone marrow in only 30-40% of cases to the onset, progress to 80-90% in advanced stages of the disease. This process can also occur in organs belonging to the reticuloendothelial system (lymph nodes, spleen, liver), and occasionally in cerebrospinal fluid, skin, and fluids.
Due to its clinical manifestations, it should be included in the differential diagnosis of fever of unknown origin, acute receptor failure or hepatitis with coagulopathy, sepsis with multiple organ failure, lymphocytic meningitis, or encephalitis with focal CNS lesions.
In 1991 the Histiocyte Society published a new diagnos criteria based on clinical, laboratory and histopathological alterations; which were modified in the 2004 revision to include three additional criteria based on immune response and NK cell activity; requiring at least 5 of the eight criteria that are shown in Table 1 [9]
Table 1. Diagnostic criteria for hemophagocytic syndrome established by the Histiocyte Society.
The diagnosis is established if 1 or 2 of the following are true:
- Molecular diagnosis consistent with SHF
- Diagnostic criteria for SHF (At least 5 of the following 8 :)
|
CLINICAL CRITERIA |
Fever |
>7 days, ≥ 38.5°C |
Splenomegaly |
(≥ 3 cm) |
LABORATORY CRITERIA |
Cytopenia (≥2 or 3 cell lines in peripheral blood) |
Hemoglobin
Platelets
Neutrophils |
<9 g/dl
<100,000 xul
<1,000 u/l |
Hypertriglyceridemia and/or Hypofibrinogenemia |
> 265 mg/dL or> 3 mmol/L
<1.5 g/l |
Hyperferritinemia |
> 500 ug/l |
Decreased or absent NK cell activity |
Soluble CD25 |
> 500 ug/l |
HISTOLOGICAL CRITERIA |
Hemophagocytosis in the bone marrow, spleen, or lymph nodes |
In 2009 Filipovich AH modified diagnostic criteria to assist with a quick diagnosis and opportunistic treatment (Table 2) [10].
Table 2. Proposed HLH diagnostic criteria
1. Molecular diagnosis of hemophagocytic lymphohistiocytosis (HLH) or X-linked lymphoproliferative syndrome (XLP). |
2. Or at least 3 of 4: |
a. Fever |
b. Splenomegaly |
c. Cytopenias (minimum 2 cell lines reduced) |
d. Hepatitis |
3. And at least 1 of 4: |
a. Hemophagocytosis |
b. ↑ Ferritin |
c. ↑ sIL2Rα (age based) |
d. Absent or very decreased NK function |
4. Other results supportive of HLH diagnosis: (Not required) |
a. Hypertriglyceridemia |
b. Hypofibrinogenemia |
c. Hyponatremia |
Treatment
The treatment is focused on suppressing the inflammatory state correction cytotoxicity. In the primary forms, the only treatment that corrects the disease is hematopoietic stem cell transplantation. The therapy implemented according to the HLH-94 and HLH-2004 protocols is based on the use of corticosteroids, immunosuppressive drugs, cytostatic drugs, immunomodulators, monoclonal antibodies and cytokine therapy [11].
Supportive treatment is required in the first instance since its presentation is often life-threatening. Includes the use of prophylactic, antifungal and sometimes antiviral antibiotics, immunoglobulins (0.5 g/kg) every 4 weeks. Protective treatment of the gastric mucosa should also be given and in the event of neurological symptoms or alterations in the cerebrospinal fluid, the use of intrathecal immunomodulatory treatment is recommended.
Regarding the initial treatment according to the HLH-2004 protocol, it is divided into 2 phases. With an initial phase (induction) consisting of 8 weeks of treatment using dexamethasone and etoposide, followed by continuation therapy in cases of persistent primary or secondary disease, under the following scheme (Table 3).
Table 3. Study result
Date |
Study report |
31.01.2020 |
Glucose 93 mg/dL, urea 111 mg/dL, BUN 52 mg/dL, creatinine 4.4 mg/dL, BT 4 mg/dL, BD 3.6 mg/dL, BI 0.4 mg/dL, ALT 146 IU/L, AST 68 IU/L, Amylase 130, DHL 1010, Coagulation test: TP 13.4 sec, INR 0.9, TPT 28.6, fibrinogen 788 mg/dL |
04.02.2020 |
alpha-fetoprotein 0.72 ng/mL, carcinoembryonic antigen 1.47 ng/mL, CA 125 40.60 U/mL, CA 19-9 5.50 U/mL. Serum creatinine 4 mg/dL, urinary creatinine 25 mg/dL, volume 3580, volume Munito 2486 ml/min, creatinine clearance 15.5 ml/min.
Uroculture: Enterococcus gallinarum >100000 CFU/mL. ViralPanel: Ag Hbs Negative, HCV negativ or , HIV negative |
05.02.2020 |
12.02.2020: Total iron binding capacity 279%, Saturation 82.1%, Transferrin 187.6, iron 229, Ferritin 1020. Creatinine 4 mg/dL, uric acid 7 mg/dL, cholesterol 268 mg/dL, HDL 20 mg/dL, LDL 163 mg/dL, VLDL 85 mg/dL, Triglycerides 427 mg/dL, BT 5.8 mg/dL, BD 4.8 mg/dL, BI 1 mg/dL, ALT 62 IU/L, AST 59 IU/L, alkaline phosphatase 279 IU/L, DHL 569 IU/L, sodium 133 mmol/L, potassium 5.2 mmol/L, chlorine 95 mmol/L, phosphorus 5.7 mg/dL, calcium 8.7 mg/dL Blood count: Hb 8.42 g/dL, Hct 26.4%, platelets 27 000/mcl, leukocytes 900, neutrophils 170, lymphocytes 680, monocytes 44, eosinophils 0, basophils 11. Direct negative coombs. Clotting times: TP 13.1 sec, INR 0.9, TPT 26, fibrinogen 780 mg/dL. |
1 8.02.2020 |
Glucose 126 mg/dL, Urea 151 mg/dL, BUN 70.5 mg/dL, Creatinine 2.3 mg/dL, Uric Acid 3.1 mg/dL, Cholesterol 132 mg/dL, Triglycerides 225 mg/dL, BT 5.6 mg/dL, BD 4.8 mg/dL, BI 0.8 mg/dL, total protein 3.6 g/dL, albumin 2.1 g/dL, ALT 63 IU/L, AST 24 IU/L, alkaline phosphatase 73 IU/L, DHL 156 IU/L, phosphorus 3.4 mg/dL, calcium 7.5 mg/dL, magnesium 2.4 mg/dL. Hematic biometry: Hb 7.72 g/dL, Hct 23.4%, platelets 11 000/mcl, leukocytes 400, neutrophils 240, lymphocytes 140, monocytes 10, eosinophils 0, basophils 4. Clotting times: TP 16.8 sec , INR 1.2, TPT 24 , fibrinogen 600 mg/dL. |
07.02.2020 |
Cervical node biopsy: Neoplastic lymphoproliferative process compatible with non-Hodgkin's lymphoma |
02.25.2020 |
Bone marrow aspirate without the presence of neoplastic cells orhemophagocytosis |
02/14/2020 |
Immunophenotype reporting CD 20+ positive 3+ in 100%, CD 10 positive 3´+, BCL2 positive 3+, BCL6 negative, TdT positive 3+. Pre-B lymphoblastic leukemia. |
31.01.2020 |
USG upper abdomen: Liver with increased diameter, heterogeneity, enlarged spleen size 16.1×9.4 cm. |
05.02.2020 |
TAC neck chest abdomen inflammatory neck adenopathy, bilateral, homogeneous, greater than 25×14, and paraortic lymphadenopathy of 14×11 mm, hepatosplenomegaly |
Initial treatment
- Dexamethasone, 10 mg / m 2 for 2 weeks, followed by 5 mg/m2 for 2 weeks, 2.5 mg/m2 for 2 weeks, 1.25 mg/m2 for 1 week, and discontinue.
- Etoposide, 150 mg/m2, twice weekly for weeks 1 and 2, followed by a weekly dose for weeks 3 to 8.
Follow-up treatment
- Pulses of dexamethasone 10 mg/m2 for 3 days every 2 weeks.
- Alternating pulses of dexamethasone with etoposide 150 mg/m2.
- Cyclosporin A to from week 9 keeping average blood levels of 200 mg/L.
If patients persist without at least partial improvement in symptoms after 2-3 weeks, rescue therapy should be started. Among the drugs that are included in rescue therapy are infliximab, alemtuzumab, anakinra, and vincristine [8].
Monitoring of response to treatment is based on physical examination and laboratory studies to determine liver and kidney function, clotting times, and cytopenia.
Allogeneic transplantation is performed in patients with familial hemophagocytic syndrome, in recurrent or progressive diseases, or in those with alterations in the central nervous system. After this treatment, a 5-year post-transplant survival is estimated to be 50-65% [8].
Female or 23 years without relevant history, begins his illness with jaundice, abdominal pain, epigastric, pain, loss of weight, headache, arthralgia and myalgia. The patient was hospitalized with suspicion of Weil Syndrome´s receiving management with dicloxacillin and blood transfusion because of pancytopenia, later discharged due to partial improvement. Later she developed metrorrhagia and cervical lymphadenopathy, sent to our unit again with sever pancytopenia, acute kidney failure, and tumor lysis syndrome. The physical examination revealed conjunctival jaundice, the left infra-mandibular region with well-defined palpable adenopathy, micro vesicular dermatosis on the anterior aspect of the thorax, and hepatosplenomegaly. He received management in the intensive care unit with fluid management, anti-hyperkalemic measures, colony stimulating factor, broad spectrum antibiotic coverage and transfusion of blood products as support measures. Polyculture was performed, with development of urine culture with enterococcus gallinarium adjusting antibiotic management according to antibiogram. The tomographic scan reported inflammatory lymphadenopathy in the neck, retroperitoneum, and hepatosplenomegaly, for which a cervical node biopsy was performed with report of a neoplastic lymphoproliferative process compatible with non-Hodgkin's lymphoma, sending immunohistochemistry for differentiation, where they reported pre-B lymphoblastic leukemia, starting steroid management. The patient continued with fever and acute renal injury despite management, it was performed renal replacement therapy with hemodialysis and bone marrow aspirate reporting absence of neoplastic cells and absence of Hemophagocytosis. The patient begans with abdominal pain it was made exploratory laparotomy, finding bilateral grade II salpingitis, endometriosis and perforated cecal appendix, presenting multiple complications with hemodynamic deterioration, dehiscence, and infection of the surgical wound and persistence of pancytopenia refractory to management with blood products. The patient developed septic shock and died 2 days later in the ICU unit.
A case of a young female patient with a febrile syndrome approach is presented. According to clinical presentation consisting of persistent fever, jaundice, abdominal pain and hepatosplenomegaly and pancytopenia, also in the context of a city in which is an endemic disease raised the suspect of leptospirosis, it was started treatment without improvement, then added palpable lymphadenopathy, menorrhagia, progressing to acute renal injury and then renal failure, acute liver failure, for which we proceeded to rule out other infectious agents (HIV infection, viral hepatitis, toxoplasmosis, cytomegalovirus, rubella, syphilis), autoimmune processes and neoplasms, it was taken a biopsy of a cervical node with Immunohistochemistry with positive staining CD 20, HE, TDT, CD5 reporting pre-B lymphoblastic leukemia as a diagnosis (Figure 1).
When questioning, the patient denies a history of hematological diseases, cancer, exposure to oncogenic agents, or smoking. Despite the fact that certain clinical characteristics were compatible with leukemia, the condition did not completely justify the presentation and clinical evolution, so an analysis of the case was carried out, determining that it met the 5 criteria proposed by the HLH-2004 for hemophagocytic syndrome (Hyperferritinaemia, hypertriglyceridemia, fever, splenomegaly and pancytopenia), also the criteria proposed by Filipovich in 2009, concluding the diagnosis of hemophagocytic syndrome despite not documenting hemophagocytosis in the biopsy sample, however it was described in a large percentage of patients that it is not possible to find this lesion because Hemophagocytosis may not be clearly apparent in the initial bone marrow biopsy early in the disease process. When performing the bone marrow aspirate, no neoplastic cells were found, although it is suspected that the result would have been modified by the previous administration of steroids (Table 1).
Regarding the limitation in the diagnosis of this entity, a lack of suspicion and knowledge is suggested, generating a delay in the diagnosis with an impact on mortality. In relation to its multifactorial etiology, it is essential that the physician keep this entity in mind within the differential diagnoses, to request the pertinent studies that allow an early diagnosis and early treatment.
Hemophagocytic syndrome is a rare immune disease, in relation to its etiology it is multifactorial, is essential to physician to keep this entity in mind within the differential diagnoses in a patient with prolonged fever, cytopenia’s, hepatosplenomegaly, and suspected hemophagocytosis and to guide the diagnosis with the pertinent studies that allow an early diagnosis and prompt treatment.
- Gonzales L, Fernández A, Molina B, Bacilio C (2017) Síndrome Hemofagocítico secundario a infección por Citomegalovirus post trasplante renal. Reporte de caso. Rev Med Herd 28: 182-186.
- Janka GE (1983) Familiar hemophagocytic lymphohistiocytosis. Eur J Pediatr 140: 221-230.
- Manuel Ramos C, Pilar Brito Z, Armando López G, Munther AK, Xavier B (2014) Adult haemophagocytic syndrome. Lancet 383: 1503-1516. [Crossref]
- Dávila Dupont D, De la Peña López I (2019) Síndrome hemofagocítico. Reporte de un caso y revisión de la literatura. Rev Fac Med UNAM 62: 15-21.
- Robin D, Jeanne S, Thomas P, Bruno D, Laurent S, et al. (2003) Reactive hemophagocytic syndrome in adult systemic disease: Report of twenty-six cases and literature review. Arthritis Rheum 49: 633-639. [Crossref]
- Astigarraga I, González Granado L, Allende LM, Alsina L (2018) Haemophagocytic syndromes: The importance of early diagnosis and treatment. An Pediatr 89: 124.e1-124.e8. [Crossref]
- Espinosa Bautista KA, Garciadiego Fossas P, León Rodríguez E (2013) Síndrome hemofagocítico Conceptos actuales. Gac Méd Méx 149: 431-437.
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, et al. (2007) HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48: 124-131. [Crossref]
- Filipovich AH (2009) Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program 2009: 127-131. [Crossref]
- Paul La R, AnnaCarin H, Melissa H, Tatiana von Bahr G, Rafal M (2019) Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 133: 2465-2477. [Crossref]
- Al-Samkari H, Berliner N (2018) Hemophagocytic Lymphohistiocytosis. Annu Rev Pathol Mech Dis 13: 27.
Editorial Information
Editor-in-Chief
Andy Goren
University of Rome, Italy
Article Type
Case Report
Publication history
Received date: January 21, 2021
Accepted date: January 28, 2021
Published date: February 04, 2021
Copyright
©2021 Yessica L. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
©2020 Yessica LC, Andres AS, Luis DCO, Cecilia CA, Carlos Ivan CM, et al. (2021) Case report of hemophagocytic syndrome complicated by septic shock. Clin Case Rep Rev, 7: DOI: 10.15761/CCRR.1000495.