Abstract
Cardiac structural damage, significant arrhythmogenesis and contraction disturbance are generated by brain disorders. Recent findings cast new light on these phenomena, exposing brain areas intimately concerned with cardiac representation and function these also sub serving decision-making and social interactions. The pivotal role of the insula will be detailed, regulatory effects on cardiac sympathovagal balance discussed and ensuing effects on modification of cardiomyocyte ionic channels, and calcium economy outlined. How these lead to significant cardiac repolarization changes, arrhythmias as well as cardiac damage will be explored; such considerations may provide fresh insights into investigation and management of patients with idiopathic and intractable arrhythmias and cardiomyopathy.
Introduction
Following Schiff’s discovery in 1875 of heart rate changes on motor cortex stimulation, responses were also elicited from the frontal lobes, subcallosal gyrus, septal area, temporal pole, and cingulate gyrus [1]. Subsequently, ECG changes were produced on feline sigmoid, anterior Sylvian and extosylvian gyri stimulation [1]. In 1926 Burdenko reported that midbrain lesioning in dogs generated scattered subendocardial hemorrhages, and focal myocyte disintegration [1]. In 1933, Neuberger identified similar post-mortem changes (subsequently termed myocytolysis) in seizure patients [1].
Recently specific cardiac representation was demonstrated within the insular cortex. In the human, the insula lies within the Sylvian fissure beneath the frontoparietal operculum and the superficial temporal plane (Figure 1) occupying 2% of the total cortical area. It is a site of visceral and autonomic representation. Cardiac accelerator or decelerator effects identified on rat insular stimulation [2] are mediated by sympathetic or parasympathetic neural tone changes without adrenal involvement [2,3]. Additionally, human right anterior insular cortex stimulation elicits cardiac acceleration alone or accompanied by a pressor response, whereas left anterior insular stimulation, results in cardiac deceleration alone or accompanied by a depressor response [4]. Lesions confined to the right posterior insula increase heart rate and blood pressure, whereas left insular lesions, increase baroreceptor gain [5] indicating that the right insula regulates cardiac and vascular sympathetic tone, and the left, cardiac parasympathetic function. Furthermore, rat insular stimulation generates PR and QT prolongation, ST depression, QRS broadening, succeeded (with continuing stimulation) by ventricular bigeminy, progressive bradycardia, complete heart block and finally asystole [6] with myocytolysis apparent on autopsy (Figure 2). These features suggest sympathetic and parasympathetic coactivation, with sympathetic effects exerted on the ventricular myocardium, altering the characteristics of repolarization and producing myocytolysis, and parasympathetic activation suppressing the SA node and implementing a permissive effect on ventricular automaticity. Further lateralization effects were shown in a murine model where left (but not right) insular lesions significantly decreased myocardial contractility and were accompanied by myocytolysis [7], with a direct inverse relationship between insular lesion volume and contractility reduction.
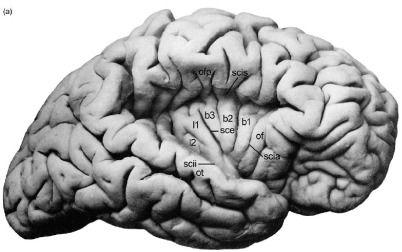
Figure 1. Human Insular Anatomy: The overlying opercula have been retracted exposing the five principal insular gyri; of is the orbitofrontal operculum; ofp is the frontoparietal operculum; ot is the temporal operculum; scia, circular insular sulcus-anterior; scii, circular insular sulcus inferior; scis, circular insular sulcus superior.
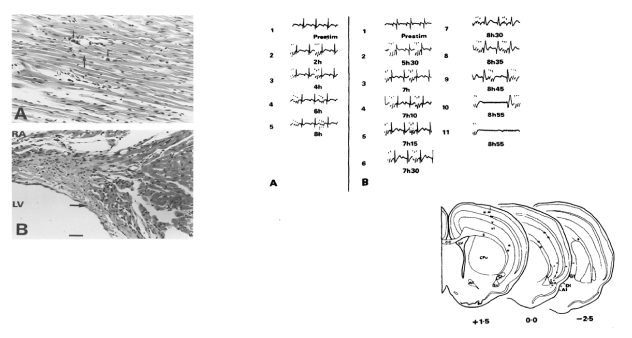
Figure 2. ECG changes and Myocytolysis on Rat Insular Cortex Stimulation: Upper Two Panels: A) coronal section through rat left ventricle demonstrating myocytolysis following insular stimulation: scattered foci of myocardial fibre disruption (thick arrows) and surrounding monocytic infiltration (thin arrows); B sagittal section of the membranous septum adjacent to the left ventricle (LV) and inferior to the right atrium (RA) showing a large subendocardial hemorrhage (thick arrow) close to the origin of the left branch of the bundle of His.
The first clinical reports [8] documented an association between acute subarachnoid hemorrhage (SAH) QT prolongation and tall peaked or inverted T waves (Figure 3) mimicked by changing left ventricular endocardial temperature. Autopsy detailed cardiac pathology like that reported earlier by Neuberger. Termed “myocytolysis”, the changes are seen in conditions where increased sympathetic tone predominates [9,10]. The histopathological spectrum was subsequently amplified to include focal areas of myocardial necrosis and inflammatory response scattered throughout the ventricle, but especially in the bundle of His (Figure 4); these are not centered on blood vessels but on nerves. Regarding cerebrogenic ECG findings and confirming their non-ischemic origin, Kono et al. [11] separated SAH patients into two groups: those with and those without repolarization changes, all lacking cardiac history. On angiography, the coronary arteries of those with ECG changes were completely normal. Abildskov et al. (cited in 1) proposed that cerebrally-related changes primarily affect cardiac repolarization and since they are mimicked by stellate ganglion stimulation are of sympathetic origin.
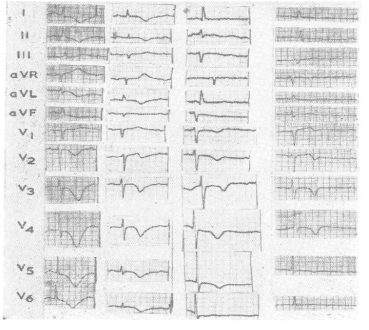
Figure 3. ECG changes and Intracerebral Hemorrhage: Four cases of subendocardial hemorrhages, and absence of coronary arterial disease. Note characteristic cerebrogenic repolarization abnormalities: QT prolongation, T wave inversion in the lateral leads, U waves.
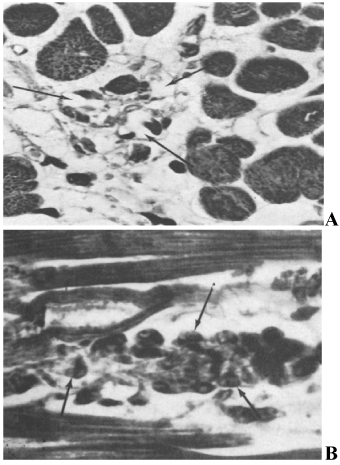
Figure 4. Myocytolysis: A. Loss of myocardial cells (arrows) and collapse of supporting stroma following SAH; B: Necrotic myocardial cell with myofibrillar degeneration and infiltration of the necrotic debris with mononuclear cells (arrows) following SAH. Lower Panels: A) Serial ECGs during control rat sensorimotor cortex stimulation without significant changes. Stimulus artifact is identified prior to the QRS complex; B) serial ECGs during insular microstimulation showing: progressive ST depression, evolution of the U wave, QT interval prolongation and progressive degrees of heart block. Subsequently, interventricular conduction defects are noted, bradycardia with a ventricular escape rhythm, complete heart block and then asystole. Inset is a coronal section diagram through the rat forebrain showing insular stimulation and control sites. Filled circles indicate insular stimulation sites; open squares indicate sites of electrode insertion without stimulation; stars indicate control stimulation sites in extra-insular locations.
Clinical expression
The cardinal signs of cerebrogenic cardiopathy (ECG repolarization changes, cardiac arrhythmia, structural damage and contraction abnormalities) manifest after stroke, during and after seizures and with emotional stress. All can be associated with sudden cardiac death. Insular involvement may play a key role in determining these outcomes.
Stroke
QT prolongation, ST elevation/depression, and U waves are specifically associated with acute SAH or intracerebral hemorrhage, but also (with lesser incidence) after ischemic stroke [12]. Comparing pre-stroke with admission ECG, new onset cardiac repolarization changes were identified in 74% compared with 14% of controls. There was no relationship to potassium levels or stroke occurring in the embolic setting of an acute myocardial infarction; 8% showed new onset ventricular fibrillation, although since this was derived from isolated ECG and not Holter data, it likely underestimates the incidence. Recently, ventricular arrhythmias were reported in 22% patients admitted without cardiac history and suffering their first stroke (a confounding issue clearly relates to the shared risk factors for stroke and cardiac ischemia) [13]. These were more common when comparison was made with TIA patients (sharing the same risk factors for cardiac ischemia) and especially so in those with right hemisphere stroke. Additionally, since the arrhythmias disappeared within 2 weeks a probable association occurred with the stroke itself rather than an underlying cardiac cause. Peaked or inverted T waves, ST segment elevation/depression were identified in 24% of acute ischemic strokes [14] with ectopy more frequently encountered compared with controls. This persisted when excluding patients with cardiac history or previous stroke. Regarding repolarization inhomogeneity, QT dispersion is increased after acute stroke compared to age-matched hospitalized controls, both groups without history or clinical evidence of cardiac disease [15]. Since this was noted only within 24 hours of stroke onset, it strengthens the association and interestingly, was noted specifically with insular infarcts [16]. Further insular association arises from the linkage between right insular stroke and AV block, QTc prolongation, T wave inversion and ventricular ectopy, (these not observed with strokes in other locations) and with increased mortality at 3 months [17,18].
Myocytolysis frequent after SAH also occurs after ischemic stroke (Smith cited in 2), albeit less frequently. Treating SAH patients with propranolol and phentolamine [10] abolished ECG repolarization changes and myocytolysis, this implicating cerebrally-derived sympathetic dysregulation in the etiology. These post-mortem observations have a pre-mortem expression in cardiac enzyme elevation indicative of structural damage. Troponin I is elevated in 35% of acute (specifically left hemisphere) strokes [19] correlating with clinical severity [20]. A further association with T wave inversion and ST depression as well as cardiac death has been reported [21]. In other studies, right insular infarction associated with Troponin T elevation, ST depression and Q waves [22] correlating with ventricular contractility impairment.
Contractility disturbance is also a pre-mortem manifestation of stroke-related cardiac structural damage. Apical akinesia with T wave inversion occurs in SAH patients lacking cardiac history or examination [23] (Figure 5) and in ischemic stroke with an incidence of 1.2% [24]. Interestingly, the majority (nearly 70%) associates with left insular infarction. Whereas these changes are evanescent, disappearing after a few days, persistence for several months has been reported [25] and is a harbinger of ventricular arrhythmias and sudden cardiac death [26].
A 1.5% annual incidence of sudden cardiac death after ischemic stroke is reported specifically associated with left hemisphere involvement, this still holding after adjustment for concomitant heart disease and medication [27]. In contrast, all cause as well as vascular mortality is increased specifically after right insular infarction and correlates with QTc prolongation and incident non-sustained ventricular tachycardia [28]. In other studies, no association between insular infarction and mortality was identified [29]. Focusing these data by removing symptomatic coronary artery disease (SCAD) patients, identified a specific association between left insular infarction and adverse cardiac outcomes and cardiac contractility reduction. Since the incidence of beta-blocker treatment was higher in SCAD patients this likely mitigated the cardiac effect of left insular stroke; additionally, symptomatic cardiac disease could subordinate the effect of insular location on cardiac outcomes; finally, ischemic preconditioning in those with symptomatic disease may be protective against cerebrally-derived cardiac mechanisms.
Sympathetic mechanisms contribute to the regulation of the QT interval [30] and are involved in the stroke-related phenomena described. Catecholamine levels are elevated after ischemic stroke [31] especially when involving the right insula. A shift to sympathetic predominance within 3 days of stroke onset [32] associated with VT deteriorating into VF and an 11% sudden death rate (mostly with right insular involvement) has been reported.
Stroke can therefore cause cardiac repolarization changes, arrhythmias, damage and contractility disturbance these contributing to mortality. There is an association with insular location although lateralization is controversial possibly related to study differences in insular infarct volume, inclusion of patients with previous stroke and/or areas of involvement beside the insula, manner of treatment of concomitant cardiac disease, differences in endpoints and their ascertainment and the modifying effect of medication. When observed, shifts towards increased sympathetic tone likely underlie these cerebrogenic effects.
Stress
Stress perception is complex generally requiring cortical involvement. Historical reports identified significant cardiac arrhythmia onset in startled patients without concomitant cardiac disease [33,34]. In a study of normal individuals in a stressful environment ventricular ectopy developed in the majority associated with increased catecholamine levels and abolished by beta blockers [35]. In patients with known heart disease, the frequency and severity of ventricular arrhythmia [36] increased with stress intensity and reduced with meditation or beta blocker treatment. Mental stress increases QT interval independent of an effect on heart rate (kept constant by pacing) while also increasing cardiac sympathetic tone, indicating a specific effect on the ventricular myocardium [37]. Consequently, stress-related increased plasma catecholamine levels are associated with T wave alternans (a measure of repolarization instability and a predictor of VF and SCD) [38]. In patients with cardiomyopathy this propensity also predicts the occurrence of ventricular tachyarrhythmias over 3 year follow up [39]. Clinically, sudden cardiac death is associated with stressful psychological events [40]: when compared to myocardial infarction uncomplicated by an arrhythmia, those resuscitated from idiopathic ventricular fibrillation are significantly more likely to have experienced acute stress in the preceding 24 hours [41].
The threshold for ventricular extrasystole induction is significantly reduced when dogs are placed in a stressful environment [42] an effect reproduced by left stellate ganglion stimulation or catecholamine infusion and abolished by beta blockade indicating mediation by sympathetic mechanisms. In pigs [43] a forebrain-originating pathway is involved in the development of stress-related VF and implicates cortical involvement. Rabbit studies [44] indicate an insular role, and human functional MRI studies have shown that subjects with an exaggerated cardiac response to stress had significantly enhanced insular activation compared with those manifesting a blunted response [45].
Takotsubo cardiomyopathy presents with significantly reduced ejection fraction, chest pain and dyspnea during a period of severe mental anguish [46]. Coronary angiography is frequently normal and there is often no cardiac history. Repolarization changes that accompany the echocardiographic findings indicate a cerebrogenic origin: T wave inversion, ST segment elevation, prominent U waves, and QTc prolongation. Cardiac enzymes are elevated and there is reduction of apical contractility. Although the prognosis is often benign, cases associated with sudden cardiac death are reported. Plasma catecholamines are elevated and reach levels higher than those seen with myocardial infarction. Additionally, increased norepinephrine levels have been found on direct sampling of coronary sinus blood [47]. This cardiomyopathy can be recreated in the rat during immobilization stress and prevented by pretreating the animals with sympathetic blockers [48]. Coronary vasodilators are without effect further indicating the absence of coronary spasm. Takotsubo-like changes have particularly been noted after left insular stroke implicating this region in the cardiopathy [49] (Figure 5).
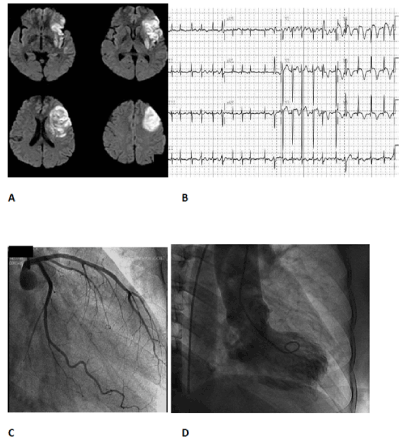
Figure 5. Insular infarction and Takotsubo cardiomyopathy: A) MRI showing left insular infarction; B) admission ECG showing atrial fibrillation and neurogenic changes comprising T wave inversion, QT prolongation, ST elevation predominantly in the anterolateral leads; C) normal coronary angiogram; D) ventriculograph typical of Takotsubo cardiomyopathy, with the apex of the heart showing the rounded appearance contrasting with the systolic constriction of the left ventricular base.
Epilepsy
Seizures provide a natural model for studying the interface between cerebral and cardiac events. SUDEP (sudden unexpected death in epilepsy) is a clinically relevant aspect. Finally, animal seizure models have added significantly to our understanding of the mechanisms underlying cerebrogenic cardiopathy.
SUDEP occurs during/following a seizure, is unrelated to status epilepticus (SE), accident or other identifiable cause [50] has an incidence of 1/150 (24 times that in the general population) and is associated with bradyarrhythmias and asystole [51,53]. Video-EEG telemetry shows a propensity to right focally- onsetting temporal lobe seizures with 8% being of insular origin [52] (however many temporal lobe seizures are probably of insular onset). Respiratory abnormalities are documented implying that SUDEP is a primary apneic event with cardiac effects being secondary; yet no consistent association between ictal respiratory changes and bradyarrhythmias/asystole has been conclusively demonstrated [53]. In the non-SUDEP setting, cardiac dysrhythmias are documented with seizures originating in the insula, mesial temporal lobe, amygdala and marginal gyrus (reviewed in 1) frequently in those without concomitant heart disease. Ictal bradycardia is reported with left temporal (possibly insular) foci [51,53].
An association between right temporal lobe seizures and QTc prolongation has been described [54], suggesting an effect on ventricular depolarization independent of heart rate. Bundle branch block, QT prolongation, T wave changes and first degree atrioventricular block were found in 35% of epileptics without evidence of underlying cardiac disease [55]. Prolonged QRS duration, incomplete right bundle branch block, interventricular conduction delay, and a higher incidence of early repolarization is reported in SUDEP patients, compared with those not succumbing to this outcome [56]. The increased interventricular conduction time is considered to reflect fibrosis secondary to chronic myocytolysis. QT and QTc dispersion are increased in epileptics this being independent of medication [57,58]. Nearly 50% of epileptics may have late potentials, unrelated to cardiac history or electrolyte imbalance a possible reflection of seizure-induced changes in cardiac ionic channels (see below) [59]. T wave alternans and asymptomatic ST segment depression during partial seizures have been described [60,61]. This may reduce the threshold for ventricular arrhythmia onset as shown by Bealer et al. [62] in a rat seizure model.
Regarding cardiac pathology, Manno et al report myocytolysis in 73% patients dying following SE [63] and this is also noted in SUDEP [64]. The likely pre-mortem expression of this pathology, Takotsubo cardiomyopathy, has been observed after seizures and is associated with QT prolongation and T wave inversion: endocardial biopsy in these cases is consistent with myocytolysis with normal cardiac arteries present on angiography. Cardiac MIBG-scanning is consistent with increased cardiac sympathetic neural activity [65,66].
As mentioned, SUDEP manifests mainly with bradyarrhythmia and asystole and this has been shown not surprisingly to be associated with increased cardiac parasympathetic tone [67]. In temporal lobe epilepsy (TLE), MIBG studies show reduced cardiac sympathetic innervation [68] unrelated to co-incident cardiac disease or medication. Chronically, increased sympathetic tone secondary to recurrent seizures is considered to cause transsynaptic degeneration of sympathetic neurons resulting in the demonstrated decreased MIBG uptake. Furthermore, when compared to TLE patients without bradycardia TLE patients with seizure-related bradycardia and asystole have significantly reduced MIBG uptake [69]. Loss of peripheral cardiac sympathetic innervation likely leads to unopposed parasympathetic tone, which becomes pathological during ictal episodes and subsequently may manifest as SUDEP.
Epilepsy models afford an opportunity to link cerebral events with autonomic activity. In cats with induced seizures, T wave flattening, U wave generation, QRS notching, Q waves, QT shortening or lengthening and ST elevation/depression were always preceded by spike activity, these changes normalizing once the spikes ceased [70]. As the induced spike frequency was increased, ventricular tachycardia occurred followed finally by asystole and death. At onset, spike activity was accompanied by synchronous firing in cardiac sympathetic and vagus nerves. Baroreflex inhibition occurred but only during cerebral spiking. As spike activity increased, desynchronization developed between different individual sympathetic nerve branches and between these and the vagus. This loss of synchronicity was suggested to induce geographical changes in ventricular myocardial excitability and consequent ventricular arrhythmia. At the highest induced spike frequencies, vagal activity markedly increased whereas cardiac sympathetic activity sharply decreased accompanied by bradyarrhythmia and subsequent asystole. In other studies, Little et al [71] showed that atenolol markedly decreased the incidence of epilepsy-associated QTc prolongation and reduction of left ventricular contraction, indicating an accompanying sympathetic component. It is therefore noteworthy that seizure-related effects can be: temporal, with desynchronized firing between various cardiac neural branches; geographic, with inhomogeneity of effect location throughout the ventricular myocardium; and reflex, with reduced BRS arising from reduced capture of vagal neural activity by peripheral blood pressure changes. Chronic cardiac stimulation related to epileptic discharge, with augmented parasympathetic and sympathetic tone and the inhomogeneity of ventricular repolarization reflected in QT dispersion, together with the changes that chronic sympathetic neural activity imposes on ion channels and cardiomyocyte calcium economy (see below) provide ample grounds for the arrhythmic basis of epileptic cardiopathy.
SUDEP is not a rare event and is associated with bradyarrhythmias and asystole and with augmented parasympathetic tone. Evidence suggests left hemisphere predominance and possibly insular origin. Other seizure-related cardiopathic effects, including myocytolysis, certain ECG changes and contractility disturbance may be of sympathetic origin. Not all patients are susceptible to ECG changes, arrhythmias or SUDEP and other factors may be involved; an association with congenital or acquired cardiac ionic channelopathies has been suggested but not conclusively confirmed [72].
Mechanisms
Dysregulatory cerebral states (especially those involving the insula) produce a tilt towards increased cardiac sympathetic tone, with the addition that in epilepsy significant augmented parasympathetic effects also occur. Recent research has increased our understanding of mechanisms whereby altered cerebrogenic states are translated into cardiac molecular and channelopathic effects thereby contributing to pathological outcomes.
Sympathetic receptor- mediated mechanisms
Many of the cerebrogenic effects discussed are mediated through sympathetic mechanisms [73]: β-Adrenergic receptors selectively couple to the adenylyl cyclase stimulatory G protein (Gs). Gs-stimulated adenylyl cyclase increases the level of cyclic adenosine monophosphate (cAMP) and activates protein kinase A. Substrates phosphorylated by protein kinase A that may be relevant to cardiac arrhythmias include the L-type Ca2+ channel (LTCC), the cardiac ryanodine receptor type 2 (RyR2), the1,4,5-triphosphate receptor (IP3R2), the α-subunit of the cardiac sodium channel, the cardiac isoform of the sodium-calcium exchanger (INa−Ca), K+ channels (IK1, HERG, and Ca2+-activated IKs),Ca2+-activated chloride current (ICl/Ca), andNa+/Ca2+ exchange current (INa−Ca). The outcome of catecholamine activation on the myocardium depends on several factors including the effect on cell type and its location, as well as catecholamine dosage. For example, α1 stimulation prolongs the action potential in Purkinje cells but decreases this in M cells. Low catecholamine doses may lengthen and high doses normalize the action potential in epicardial and endocardial cells. Torsades de pointe is an example of a sympathetic neurally-induced arrhythmia associated with the long QT syndrome (LQTS), and its occurrence is reduced by left stellate gangliectomy or by β-blockade. It is postulated that this arrhythmia is secondary to catecholamine- induced early afterdepolarizations (EADs) and that geographic variability leads to repolarization inhomogeneity setting the ground work for the arrhythmia. In a canine model of LQT1, action potential duration (APD) was prolonged homogeneously in epicardial, endocardial and mid-myocardial cells. Isoproterenol infusion resulted in torsades development with prolongation of the APD in the M cells and shortening in epicardial and endocardial myocytes. This geographic inhomogeneity (and the arrhythmia), was abolished by β-blockade.
In a rat epilepsy model, cardiac β1-receptor protein was reduced indicating chronic activation and Cx43 (connexin 43) levels (a ventricular gap junction protein which augments myocyte electrical coupling) elevated [74]. Increased phosphorylation of intracellular intermediary proteins affected by sympathetic activity was noted including protein kinase A (PKA), extracellular signal regulated kinase (ERK), calcium/calmodulin dependent protein kinase II (CaMKII) as well as decreased levels of the potassium channel Kv4.2 and HCN2 (hyperpolarization-activated cyclic nucleotide-gated channel subunit 2) whose activity is modulated by these activated kinases. Proteins responsible for regulating intracellular calcium levels and distribution were also affected including: NCX-1 (involved with Ca extrusion) which was downregulated, and a major sarcoplasmic binding protein (CaR) whose levels were elevated. Cerebral activation can therefore augment cardiac sympathetic tone (indicated by the increased kinase activity) with resulting effects on potassium channels; this may significantly affect cardiac repolarization. Additional effects on proteins which increase electrical coupling allow for the generation of arrhythmias, especially if these are not uniformly expressed throughout the myocardium. Whether similar effects occur in the human is yet unclear.
Norepinephrine induces Cx43 transcription in cultured rat cardiomyocytes [75] and consequent intercellular electrical coupling. In vivo, isoproterenol increases Cx43 expression, heart rate, and cardiac output. β-adrenergic stimulation also opens existing gap junctions and thereby enhances coupling. It was therefore suggested that any geographic variability in the distribution of such upregulation might generate inhomogeneous depolarization and the production of ventricular arrhythmias, a mechanism postulated to be operating during seizures and other cerebrogenic cardiopathic circumstances.
Potassium channelopathies- Genetic and acquired
Potassium channels contribute significantly to cardiac repolarization and are reduced in some cardiomyopathies associated with sudden cardiac death. In a rat epilepsy model increased cardiovascular sympathetic tone was associated with reduced Kv4.2 levels (a channel crucial for repolarization), increased QTc dispersion and Troponin I elevation, these effects being blocked by atenolol [76]. The exact cause of this reduction is unclear, however in vitro studies indicate that CaMKII (activated by β-receptors) phosphorylates Kv4.2 potassium channels and changes the amount present in the cell membrane [77].
It has been suggested that brain-derived effects result in cardiopathy only in the presence of a cardiac channel mutation. Contradicting this, the KCNA1 gene encoding Kv1.1 channels is found in brain and in vagal axonal juxtaparanodes, but not in heart muscle [78]. In KCNA1 knock-out mice, seizures associated with prolonged periods of sinus bradycardia, conduction block, and frequent PVCs occur. These cardiac concomitants are solely of neural origin since they are blocked by atropine. Seizures unmask parasympathetic hyperexcitability and result in bradycardia and conduction block in these KCNA1 knock-out mice as the role of this channel is to suppress backfiring following vagal nerve action potential generation. This indicates that mechanisms restricted to the neuraxis alone can cause cardiac effects without requiring an additional intrinsic cardiac channelopathy. The role of the brain as a promotor of arrhythmogenesis in the presence of an intrinsic cardiac channelopathy has also been demonstrated: LQTS1 is associated with seizures (22% of patients) and is caused by a gene mutation which encodes the alpha subunit of the tetrameric voltage-gated potassium channel KvLQT1 (Kv7.1). These mutations decrease the slow repolarizing Iks current. In a mouse KvLQT1 mutation model AF, atrial flutter, PVCs, and episodic atrio-ventricular conduction block occur but only when coupled with concomitant spikes indicating that although there is an intrinsic cardiac channelopathy a trigger in the form of an epileptic discharge is also required [79].
Sodium channelopathies- Genetic and acquired
Cardiac sodium (Na) currents include: a fast-inward current accounting for the peak of the action potential and a sustained late component (INaL). This latter is a significant contributor to QT interval duration. In a rat epilepsy model, cardiac TTX-sensitive Na channel (Nav1.1) DNA expression was increased compared to controls and correlated with QTc and myocyte action potential prolongation [80]. Cardiomyocyte membrane TTX- sensitive Na channels (which augment the INaL current) were also increased and these are more likely to be activated at a lower threshold than normal cardiac Na channels (since their activation threshold is close to the resting potential). Consequent delayed recovery from inactivation may increase the refractory period and contribute to the conduction block not uncommon in epilepsy (and in neurogenic ECG changes in general) and the bradycardia thought to underlie SUDEP.
Mention has been made of the co-occurrence of mutated ion channels in the heart and brain. Kalume et al [81] explored the possible relative contribution of each in a mouse model of Dravet syndrome, an epileptic encephalopathy associated with loss of function mutation in the SCN1A gene encoding the brain type 1 voltage gated Nav1.1 sodium channel. Dravet syndrome is accompanied by a high incidence of sudden death and since the affected channels are expressed in both heart and brain, it is unclear which is the principal culprit. Sudden death with preceding AV block occurred only after a seizure and with presence of the mutation in forebrain GABAergic interneurons, whereas cardiac-specific mutations were not associated with sudden death. Muscarinic antagonists prevented these deaths indicating a parasympathetic mechanism. The authors postulate that this is caused by hyperactivation of pathways from the forebrain to brainstem parasympathetic cardiac neurons due to loss of interneuronal inhibition.
Linking acquired Na channelopathies with increased sympathetic activation, Wagner et al [82] transfected cardiac myocytes so that CaMKII (activated by β-receptor stimulation) was overexpressed. Enhanced CaMKII phosphorylation of Na channels was found as well as concomitantly increased INaL and intracellular Na concentration. In vivo, QRS and QT intervals were prolonged, and the threshold for VT development decreased. This sympathetically- mediated CaMKII phosphorylation of Na channels resulted in slowing of fast inward Na current inactivation and enhancement of the late Ina current thereby increasing action potential duration and generating early afterpotentials (EADs). The elevated Nai because of slowed inactivation could also result in late afterdepolarization formation.
Camkii, Mitochondria and Calcium
Intracellular molecular effects mediating cerebrally-induced cardiac structural and functional changes will be discussed in this section. In short, sympathetic-mediated upregulation of CaMKIIδ (the cardiac isoform of calcium/calmodulin protein kinase) and compartmental shifts in intracellular calcium concentration seem pivotal. CaMKII has important effects on cardiac myocyte ion channel expression and activity, as well as intracellular Ca levels and likely plays a crucial part in the origin of cerebrogenic cardiopathy.
β1-receptor stimulation activates two phosphokinases [83,84]: a) phosphokinase A (PKA) mediating short -term (“flight or fright”) effects and RyR2 (sarcoplasmic reticulum ryanodine receptor) activation with consequent Ca release; b) chronic sympathetic stimulation activates CaMKII and contributes to the pathological changes that may occur with stroke, stress, and seizure. Under basal conditions CaMKII is in a state of autoinhibition but as calcium levels increase subsequent upon either sarcoplasmic reticulum (SR) release or LTCC activation, calcium complexes with calmodulin and then binds to CaMKII releasing it from autoinhibition. CaMKII regulates SR Ca uptake and release [85]. Target proteins include: phospholamban, RyR2 and Ca channels which are activated and enhance excitation/contraction coupling and heart rate responses.
In heart failure (in which there is sympathetic upregulation) phosphorylation of Kv4.3 by CaMKII reduces the repolarizing K current thereby prolonging the action potential plateau phase and consequently, the QT interval. In animal models, transgenic overexpression of CaMKIIδ increases the frequency of Cav1.2 channel opening and RyR2-mediated SR Ca release thereby prolonging the action potential. CaMKII phosphorylation of Nav1.5 channels impair their inactivation and increases intracellular Na consequently reversing the Na/Ca exchanger and increasing intracellular Ca; this contributes to the generation of afterdepolarizations [83,84] (Figure 6). CaMKII activation phosphorylates potassium channels, decreases their transcription and trafficking to the cardiomyocyte membrane. Inhibition of myocardial CaMKII augments ischemic preconditioning and reduces ischemia-reperfusion cardiac damage [83,84]. Transgenic mice with CaMKII overexpression manifest AP and QT prolongation, LVH and polymorphic ventricular tachycardia. These effects were all blocked by CaMKII inhibitors.
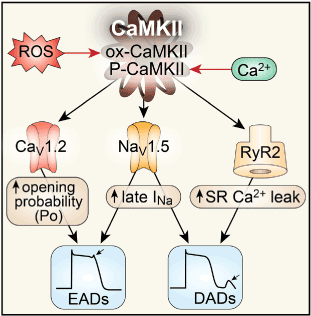
Figure 6. β-adrenoceptor activated CaMKII and the mechanism of sympathetic-induced arrhythmogenesis: CaMKII phosphorylates L-type Ca channels (CaV1.2) increasing the probability of channel opening, contributing to early afterdepolarization formation (EADs). The increased ICa thereby generated, contributes to action potential prolongation and delayed afterdepolarizations (DADs). CaMKII also phosphorylates Na+channels (Nav1.5) enhancing late INa current, promoting EADs, increasing subsarcolemmal [Na+]I promoting DAD development. CaMKII phosphorylation of the ryanodine receptor (RyR2) increases sarcoplasmic reticulum (SR) Ca2+leak, shifting the Na+/Ca2+ exchanger (NCX) to a forward mode, thus contributing to DAD formation. CaMKII contributes to the formation of injured myocardium by promoting myocyte death and collagen deposition.
Mitochondria contribute 20-30% of myocardial mass and are an important Ca buffer [85]. Excess intramitrochondrial Ca accumulation facilitates mitochondrial permeability transition pore (mPTP) opening, with loss of the mitochondrial transmembrane potential, and release of cytochrome C thus triggering apoptosis [85]. mPTP is a target of CaMKII which can also precipitate these events (Figure 7). βAR1 receptor stimulation causes ventricular myocyte apoptosis through this mechanism which is prevented by CaMKII inhibition and by β1AR antagonists. Reactive oxygen species (ROS) also derive from mitochondria due to uncoupling of the electron transport chain, and are seen in animal models of failing ventricular myocardia and in human heart failure. ROS can convert CaMKII into its autonomously active form. Countervailing protective proteins for such oxidative stress have been found: MsrA, and MsrB (methionine sulphoxide diasteroisomers) which are in the cytoplasm, nucleus and mitochondria. These reverse CaMKII oxidation, and protect against cardiac cell death in animal models. ROS generation may therefore be another mechanism contributing to cerebrogenic cardiac pathology.
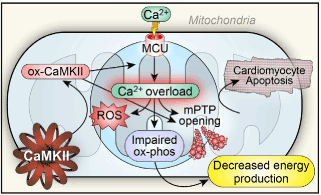
Figure 7. CaMKII mediated cardiomyocyte apoptosis-possible mechanisms underlying myocytolysis and cerebrogenic cardiopathy: the mitochondrial Ca2+ uniporter (MCU) is phosphorylated by activated CaMKII leading to excessive mitochondrial Ca2+ accumulation triggering mitochondrial permeability transition pore (mPTP) opening, and subsequent cell death. Mitochondrial Ca2+ overload also promotes ROS generation, oxidizing CaMKII (ox-CaMKII) releasing it from autoinhibition further resulting in intramitochondrial Ca accumulation and apoptosis.
In heart failure, T- tubule system structural changes occur resulting in loss of close apposition of RyR2 and Cav1.2 receptors; this impairs Ca-dependent Ca release from the SR and excitation/contraction coupling resulting in reduced cardiac contractility. Such may occur in Takotsubo and other cerebrogenic cardiopathies. Since β1AR antagonists prevent disruption of the tubular system, the sympathetic nervous system may be involved in triggering a disassociation between Cav1.2 and RyR2 receptors and consequent dysregulation of Ca release and reduced inotropy.
In summary: Through a complex interweaving of intracellular processes, sympathetic upregulation consequent upon cerebral activation can affect cardiac depolarization, repolarization, contractility and structure. Current evidence from in vitro, as well as in vivo studies and from human biopsy material suggest a pivotal role for CaMKII upregulation secondary to β1AR activation, which in turn has numerous effects on K, Na and Ca channels including determining their number and distribution within the cardiomyocyte membrane. Additional effects on the mitochondria may result in release of agents mediating apoptosis. Distribution of cardiac sympathetic nerves within the ventricular myocardium may result in non-homogenous effects targeting for example the cardiac apex as seen in Takotsubo cardiopathy or allowing the possibility of re-entry.
CAPON
CAPON (C-adaptor protein of neuronal nitric oxide synthase) is a chaperone protein modulating the effects of neuronal nitric oxide synthase (nNOS). By competing with PSD95 (an anchor protein) for nNOS it uncouples the enzyme from the plasma membrane and directs it to other sites decreasing its membrane-related activity. CAPON is present in rat cardiac sympathetic neurons and by removing nNOS from the presynaptic terminal, Ca currents (regulated by this enzyme) are reduced with concomitant reduction of norepinephrine release [86]. Postsynaptically, CAPON modulates ion channel activity in cardiac myocytes where it colocalizes with nNOS, and LTCCs and with Kir3.1 channels. CAPON upregulation shortens action potential duration by inhibiting Ica currents and activating Ikr. CAPON (and co-localized NOS) has been found in the SR and mitochondria of ventricular myocytes [87] and is involved with intracellular Ca handling and consequently regulates CaMKII activity (an important intermediary in cerebrogenic cardiopathic effects). CAPON polymorphisms could therefore render individuals particularly vulnerable to sympathetically-mediated cerebrogenic effects, by both neuronal and cardiomyocyte-mediated mechanisms and in this regard, associations between CAPON polymorphisms, QT interval prolongation and sudden cardiac death have been reported [88-90]. The effect of these polymorphisms is primarily to downregulate CAPON transcription [89] thereby increasing norepinephrine release especially in conditions of augmented sympathetic tone as occurs with stress, stroke and epilepsy contributing to the cardiopathic effects detailed above.
The insula, psychological homeostasis and cerebrogenic cardiopathy
Cortical representation of cardiovascular function provokes a rethinking of autonomic nervous system function. When faced with challenging environmental situations and a range of reaction choices, discrimination to initiate the right one is vital. The James-Lange hypothesis suggests that each choice generates an associated set of autonomic concomitants which feed back into the brain (reviewed in 1). This colors the anticipated response, its ideation and its effectuation with a set of feelings. Without them, all responses would have the same validity and selection would either be random or require time-consuming ratiocination. “Psychological homeostasis” reduces cortical stress perception by correct response selection and achievement of the anticipated outcome. Recent MRI studies indicate a key role for the right insula and cardiac input in this regard (reviewed in 1). Underpinning this is the close coupling of different areas of the brain into networks, which evaluate, respond to and choose these appropriate responses. The insula selects appropriate networks, and couples the strength of the interactions between different constituent components. A by-product is that when abnormal interactions occur as in psychiatric disorders or after stroke or seizures (especially when involving the insula), abnormal cardiac regulatory activity may result. Although there is currently little evidence regarding network reconfiguration in these conditions, in focal epilepsy changes are described in network connectivity patterns (including the insula) [91,92]: interictal spikes affected the connectivity strengths between constituent network members. Similar changes could occur with other forms of aberrant cortical activity (generated by stroke or increased stress perception for example) which might dysregulate cardiac neural regulatory efferent activity resulting in the cardiopathic effects detailed in this review.
Conclusions
Cerebrogenic effects can initiate cardiopathic changes either without underlying cardiac pathology or genetic channelopathy, or coupled with them in an interactive, permissive role. The insular cortex occupies a pivotal position in this regard likely following from its integrative involvement in cognitive and decision-making processes in which afferent and efferent insular cardiovascular influences play an important role. Therefore, psychological factors including stress, and pathological processes including stroke and epilepsy especially when involving the insula (or regions with which it is networked) are important in destabilizing cardiac function leading to both structural and electrophysiological effects with the ultimate clinical expression of sudden cardiac death. These are mediated by imbalances in cardiac autonomic regulation, especially involving the sympathetic nervous system, although parasympathetic influences are also important (especially in SUDEP). At the molecular level, CaMKIIδ upregulation is central with consequent induced sodium and potassium channelopathies producing Ca, K and Na ionic flux dysregulation and alterations in their intracellular levels. Mitochondrial dysfunction and reactive oxygen species generation play a role, with subsequent impairment of excitation/contraction coupling and cardiomyocyte apoptosis leading to either the subtle pathological changes of myocytolysis and inotropic downregulation or a more flagrant cardiomyopathy (for example Takotsubo). It would be imprudent to suggest that cerebrogenic effects predominate in the general etiology of cardiac disorders, but there are instances where neurophysiological examination could be valuable. In patients with idiopathic and intractable arrhythmia or cardiac contractility disorders, as well as those with a propensity to drug-related QT interval prolongation or etiologically-unclear vulnerability to cardiac arrest, in addition to a search for the traditional genetic channelopathies, estimation of CAPON polymorphisms, and of sympathovagal tone (including BRS) may be helpful. Autonomic tone should not only be assessed under basal conditions, but also under stress (in this case psychological) since sympathetic-related mechanisms may only manifest under these circumstances. In addition, MIBG cardiac scanning may give further information as to the status of intracardiac sympathetic neurons which are affected in conditions of chronic cardiac sympathetic upregulation. Finally, functional MRI can provide an assessment of insular activity and cerebral network relationships and balance in such vulnerable patients. In addition to gathering helpful information regarding the autonomic status of such patients, these investigations can indicate appropriate supplementary interventions additional to antiarrhythmic agents. Cardiac MIBG scanning and cerebral functional MRI afford an additional means of measuring the effects of such interventions. The use of CaMKIIδ inhibitors should they become available, might be appropriate in specific instances, as may inhibitors of oxidative stress especially in situations where there is an interaction between neurological factors and genetic channelopathies (such as those where an arrhythmia is precipitated by stress). Clearly more research is needed, and this will lead to improved identification of patients at significant risk of SUDEP or sudden cardiac death and provide insight into appropriate traditional or novel means of intervention.
References
- Oppenheimer S, Cechetto D2 (2016) The Insular Cortex and the Regulation of Cardiac Function. Compr Physiol 6: 1081-1133. [Crossref]
- Oppenheimer SM1, Cechetto DF (1990) Cardiac chronotropic organization of the rat insular cortex. Brain Res 533: 66-72. [Crossref]
- Oppenheimer SM, Saleh T, Wilson JX, Cechetto DF (1992) Plasma and organ catecholamine levels following stimulation of the rat insular cortex. Brain Res 529: 221-228. [Crossref]
- Oppenheimer SM, Gelb A, Girvin JP, Hachinski VC (1992) Cardiovascular effects of human insular cortex stimulation. Neurology 42:727-1732. [Crossref]
- Zhang ZH, Rashba S, Oppenheimer SM (1998) Insular cortex lesions alter baroreceptor sensitivity in the urethane-anesthetized rat. Brain Res 813: 73-81. [Crossref]
- Oppenheimer SM, Wilson JX, Guiraudon C, Cechetto DF (1991) Insular cortex stimulation produces lethal cardiac arrhythmias: A mechanism of sudden death. Brain Res 550: 115-121 [Crossref]
- Min J, Farooq MU, Greenberg E, Aloka F, Bhatt A, et al. (2009) Cardiac dysfunction after left permanent cerebral focal ischemia: The brain and heart connection. Stroke 40: 2560-2563. [Crossref]
- BYER E, ASHMAN R, TOTH LA (1947) Electrocardiograms with large, upright T waves and long Q-T intervals. Am Heart J 33: 796-806. [Crossref]
- Greenhoot JH, Reichenbach DD (1969) Cardiac injury and subarachnoid hemorrhage, a clinical, pathological and physiological correlation. J Neurosurg 30: 521-531. [Crossref]
- Neil-Dwyer G, Walter P, Cruickshank JM, Doshi B, O’Gorman P (1978) Effect of propranolol and phentolamine on myocardial necrosis after subarachnoid haemorrhage. BMJ 2: 990-992. [Crossref]
- Kono T, Morita H, Kuroiwa T, Onaka H, Takatsuka H, et al. (1994) Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: Neurogenic stunned myocardium. J Am Coll Cardiol 24: 636-640. [Crossref]
- Goldstein DS (1979) The electrocardiogram in stroke: relationship to pathophysiological type and comparison with prior tracings. Stroke 10: 253-259. [Crossref]
- Daniele O, Caravaglios G, Fierro B, Natalè E (2002) Stroke and cardiac arrhythmias. J Stroke Cerebrovasc Dis 11: 28-33. [Crossref]
- Abboud H, Berroir S, Labreuche J, Orjuela K, Amarenco P (2006) Insular involvement in brain infarction increases risk for cardiac arrhythmias and death. Ann Neurol 59: 691-699. [Crossref]
- Afsar N, Fak AS, Metzger JT, Van Melle G, Kappenberger L, et al. (2003) Acute stroke increases QT dispersion in patients without known cardiac diseases. Arch Neurol 60: 346-350.
- Eckardt M, Gerlach L, Welter FL (1999) Prolongation of the frequency-corrected QT dispersion following cerebral strokes with involvement of the insula of Reil. Eur Neurol 42: 190-193. [Crossref]
- Christensen H1, Boysen G, Christensen AF, Johannesen HH (2005) Insular lesions, ECG abnormalities, and outcome in acute stroke. J Neurol Neurosurg Psychiatry 76: 269-271. [Crossref]
- Tatschl C, Stöllberger C, Matz K, Yilmaz N, Eckhardt R, et al. (2006) Insular involvement is associated with QT prolongation: ECG abnormalities in patients with acute stroke. Cerebrovasc Dis 21: 47-53. [Crossref]
- Christensen H, Johannesen HH, Christensen AF, Bendtzen K, Boysen G (2004) Serum cardiac troponin I in acute stroke is related to serum cortisol and TNF-alpha. Cerebrovasc Dis 18: 194-199. [Crossref]
- Hasırcı B, Okay M, Ağırcan D, Koçer A (2013) Elevated troponin level with negative outcome was found in ischemic stroke. Cardiovasc Psychiatry Neurol 953672.
- Di Angelantonio E1, Fiorelli M, Toni D, Sacchetti ML, Lorenzano S, et al. (2005) Prognostic significance of admission levels of troponin I in patients with acute ischaemic stroke. J Neurol Neurosurg Psychiatry 76: 76-81. [Crossref]
- Fure B, Bruun Wyller T, Thommessen B (2006) Electrocardiographic and troponin T changes in acute ischaemic stroke. J Intern Med 259: 592-597. [Crossref]
- Pollick C, Cujec B, Parker S, Tator C (1988) Left ventricular wall motion abnormalities in subarachnoid hemorrhage: an echocardiographic study. J Am Coll Cardiol 12: 600-605. [Crossref]
- Yoshimura S, Toyoda K, Ohara T, Nagasawa H, Ohtani N, et al. (2008) Takotsubo cardiomyopathy in acute ischemic stroke. Ann Neurol 64: 547-554. [Crossref]
- Dias V, Cabral S, Meireles A, Gomes C, Antunes N, et al. (2009) Stunned myocardium following ischemic stroke. Cardiology 113: 287-290. [Crossref]
- Syed FF, Asirvatham SJ, Francis J (2011) Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 13: 780-788. [Crossref]
- Algra A, Gates PC, Fox AJ, Hachinski V, Barnett HJM (2003) Side of brain infarction and long-term risk of sudden death in patients with symptomatic carotid disease. Stroke 34: 2871-2875. [Crossref]
- Colivicchi F, Bassi A, Santini M, Caltagirone C (2005) Prognostic implications of right-sided insular damage, cardiac autonomic derangement and arrhythmias after acute ischemic stroke. Stroke 36: 1710-1715. [Crossref]
- Laowattana S, Zeger SL, Lima JA, Goodman SN, Wittstein IS, et al. (2006) Left insular stroke is associated with adverse cardiac outcome. Neurology 66: 477-483. [Crossref]
- Diedrich A, Jordan J, Shannon JR, Robertson D, Biaggioni I (2002) Modulation of QT interval during autonomic nervous system blockade in humans. Circulation 106: 2238-2243. [Crossref]
- Meyer S, Strittmatter M, Fischer C, Georg T, Schmitz B (2004) Lateralization in autonomic dysfunction in ischemic stroke involving the insular cortex. Neuroreport 15: 357-361. [Crossref]
- Tokgözoglu SL, Batur MK, Topçuoglu MA, Saribas O, Kes S, et al. (1999) Effects of stroke localization on cardiac autonomic balance and sudden death. Stroke 30: 1307-1311. [Crossref]
- Hai H (1977) Psychogenic ventricular tachycardia. Circ Abst 55,56 III: 156.
- Lown B, Temte JV, Reich P, Regestein Q, Hai H (1976) Basis for recurring ventricular fibrillation in the absence of coronary heart disease and its management. N Engl J Med 294: 623-629. [Crossref]
- Taggart P, Carruthers M, Somerville W (1973) Electrocardiogram, plasma catecholamines and lipids, and their modification by oxprenolol when speaking before an audience. Lancet 2: 341-346. [Crossref]
- Lown B, Da Silva RA, Lenson R (1978) Roles of psychologic stress and autonomic nervous system changes in provocation of ventricular premature complexes. Am J Cardiol 41: 979-985. [Crossref]
- Nogoescu R, Dinca-Panaitescu S, Filcescu V, Ionescu D, Wolf S (1997) Mental stress enhances the sympathetic fraction of QT variability in an RR-independent way. Integ Physiol Behav Sci 32: 220-227. [Crossref]
- Lampert R, Shusterman V, Burg MW, Lee FA, Earley C, et al. (2005) Effect of psychological stress on repolarization and relationship to autonomic and hemodynamic factors. J Cardiovasc Electrophysiol 16: 372-377. [Crossref]
- Abisse SS, Lampert R, Burg M, Soufer R, Shusterman V (2011) Cardiac repolarization instability during psychological stress in patients with ventricular arrhythmias. J Electrocardiol 44: 678-683. [Crossref]
- Kamarck T, Jennings JR (1991) Biobehavioral factors in sudden cardiac death. Psychol Bull 109: 42-75. [Crossref]
- Lane RD, Laukes C, Marcus FI, ChesneyMA, Sechrest L, et al. (2005) Psychological stress preceding idiopathic ventricular fibrillation. Psychosom Med 67: 359-365. [Crossref]
- Lown B, Verrier RL, Rabinowitz SH (1977) Neural and psychologic mechanisms and the problem of sudden cardiac death. Am J Cardiol 39: 890-902. [Crossref]
- Skinner JE, Reed JC (1981) Blockade of frontocortical-brainstem pathway prevents ventricular fibrillation of ischemic heart. Am J Physiol 240: H156-H163. [Crossref]
- Powell DA, Buchanan S, Hernàndez L (1985) Electrical stimulation of insular cortex elicits cardiac inhibition but insular lesions do not abolish conditioned bradycardia in rabbits. Behav Brain Res 17: 125-144. [Crossref]
- Ginty AT, Gianaros PJ, Derbyshire SWG, Phillips AC, Carroll D (2013) Blunted cardiac stress reactivity relates to neural hypoactivation. Psychophysiology 50: 219-229. [Crossref]
- Gianni M, Dentali F, Grandi AM, Sumner G, Hiralal R, et al. (2006) Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J 27: 1523-1529. [Crossref]
- Kume T, Kawamoto T, Okura H, Toyota E, Neishi Y, et al. (2008) Local release of catecholamines from the hearts of patients with Tako-Tsubo-Like left ventricular dysfunction. Circ J 72: 106-108. [Crossref]
- Ueyama T (2004) Emotional stress-induced takotsubo cardiomyopathy: Animal model and molecular mechanisms. Ann N Y Acad Sci 1018: 437-444. [Crossref]
- Yoshimura S, Toyoda K, Ohara T, Nagasawa H, Ohtani N, et al. (2008) Takotsubo cardiomyopathy in acute ischemic stroke. Ann Neurol 64: 547-554. [Crossref]
- Hirsch LJ, Donner EJ, So EL, Jacobs M, Nashef L, et al. (2011) Abbreviated report of the NIH/NINDS workshop on sudden unexpected death in epilepsy. Neurology 76: 1932-1938. [Crossref]
- Rugg-Gunn FJ, Simister RJ, Squirrell M, Holdright DR, Duncan JS (2004) Cardiac arrhythmias in focal epilepsy: a prospective long-term study. Lancet 364: 2212-2219. [Crossref]
- Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, et al. (2013) Incidence and mechanisms ofcardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): A retrospective study. Lancet Neurol 12: 966-977. [Crossref]
- Tinuper P, Bisulli F, Cerullo A, Carcangiu R, Marini C, et al. (2001) Ictal bradycardia in partial epileptic seizures: Autonomic investigation in three cases and literature review. Brain 124: 2361-2371. [Crossref]
- Brotherstone R, Blackhall B, McLellan A (2010) Lengthening of corrected QT during epileptic seizures. Epilepsia 51: 221-232. [Crossref]
- Surges R, Thijs RD, Tan HL, Sander JW (2009) Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol 5: 492-504. [Crossref]
- Chyou JY, Friedman D, Cerrone M, Slater W, Guo Y, et al. (2016) Electrocardiographic features of sudden unexpected death in epilepsy. Epilepsia 57: e135-e139. [Crossref]
- Akalin F, Tirtir A, Yilmaz Y (2003) Increased QT dispersion in epileptic children. Acta Paediatr 92: 916-920. [Crossref]
- Dogan E, Dogan U, Yildiz G, Hakan Akilli H, Genca E, et al. (2010) Evaluation of cardiac repolarization indices in well-controlled partial epilepsy: 12-Lead ECG findings. Epilepsy Research 90: 157-163. [Crossref]
- Rejdak K, Rubaj A, GÅ‚owniak A, Furmanek K, Kutarski A, et al. (2011) Analysis of ventricular late potentials in signal-averaged ECG of people with epilepsy. Epilepsia 52: 2118-2124. [Crossref]
- Strzelczyk A, Adjei P, Scott CA, Bauer S, Rosenow F, et al. (2011) Postictal increase in T-wave alternans after generalized tonic-clonic seizures. Epilepsia 52: 2112-2117. [Crossref]
- Tigaran S, Rasmussen V, Dam M, Pedersen S, Høgenhaven H, et al. (1997) ECG changes in epilepsy patients. Acta Neurol Scand 96: 72-75. [Crossref]
- Bealer SL, Little JG (2013) Seizures following hippocampal kindling induce QT interval prolongation and increased susceptibility to arrhythmias in rats. Epilepsy Research 105: 216-219. [Crossref]
- Manno EM, Pfeifer EA, Cascino GD, Noe KH, Wijdicks EF (2005) Cardiac pathology in status epilepticus. Ann Neurol 58: 954-957. [Crossref]
- Natelson BH, Suarez RV, Terrence CF, Turizo R (1998) Patients with epilepsy who die suddenly have cardiac disease. Arch Neurol 55: 857-860. [Crossref]
- Legriel S, Bruneel F, Dalle L, Appere-de-Vecchi C, Georges J-L, et al. (2008) Recurrent Takotsubo Cardiomyopathy Triggered by Convulsive Status Epilepticus. Neurocrit Care 9: 118-121. [Crossref]
- Shimizu M, Kagawa A, Takano T, Masai H, Miwa Y (2008) Neurogenic Stunned Myocardium Associated with Status Epilepticus and Postictal Catecholamine Surge. Intern Med 47: 269-273. [Crossref]
- Jeppesen J, Fuglsang-Frederiksen A, Brugada R, Pederson B, Rubboli G, et al. (2014) Heart rate variability analysis indicates preictal parasympathetic overdrive preceding seizure-induced cardiac dysrhythmias leading to sudden unexpected death in a patient with epilepsy. Epilepsia 55: e67-e71. [Crossref]
- Druschky A, Hilz MJ, Hopp P, Platsch G, Radespiel-Troger M, et al. (2001) Interictal cardiac autonomic dysfunction in temporal lobe epilepsy demonstrated by [123 I] metaiodobenzylguanidine-SPECT. Brain 124: 2372-2382. [Crossref]
- Kerling F, Dütsch M, Linke R, Kuwert T, Stefan H, et al. (2009) Relation between ictal asystole and cardiac sympathetic dysfunction shown by MIBG-SPECT. Acta Neurol Scand 120: 123-129. [Crossref]
- Schraeder PL, Lathers CM (1983) Cardiac neural discharge and epileptogenic activity in the cat: an animal model for unexplained death. Life Sci 32: 1371-1382. [Crossref]
- Little J, Bealer S (2012) Beta adrenergic blockade prevents cardiac dysfunction following status epilepticus in rats. Epilepsy Research 99: 233-239 [Crossref]
- Aiba I, Noebels J (2015) Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med 7: 282ra46. [Crossref]
- Anderson KP (2003) Sympathetic Nervous System Activity and Ventricular Tachyarrhythmias: Recent Advances. A.N.E 8: 75-89. [Crossref]
- Brewster A, Marzec K, Hairston A, Ho M, Anderson A, et al. (2016) Early cardiac electrographic and molecular remodeling in a model of status epilepticus and acquired epilepsy. Epilepsia 57: 1907-1915. [Crossref]
- Salameh A, Frenzel C, Boldt A, Rassler B, Glawe I, et al. (2006) Subchronic alpha- and beta-adrenergic regulation of cardiac gap junction protein expression. FASEB J 20: 365-367. [Crossref]
- Bealer S, Little J, Metcalf C, Brewster A, Anderson A (2010) Autonomic and cellular mechanisms mediating detrimental cardiac effects of status epilepticus. Epilepsy Res 91: 66-73. [Crossref]
- Varga A, Yuan L, Anderson A, Schrader L, Wu G, et al. (2004) Calcium–Calmodulin-Dependent Kinase II Modulates Kv4.2 Channel Expression and Upregulates Neuronal A-Type Potassium Currents. J Neurosci 24: 3643-3654. [Crossref]
- Glasscock E, Yoo J, Chen T, Klassen T, Noebels J (2010) Kv1.1 Potassium Channel Deficiency Reveals Brain-Driven Cardiac Dysfunction as a Candidate Mechanism for Sudden Unexplained Death in Epilepsy. J Neurosci 30: 5167-5175. [Crossref]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, et al. (2009) Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med 1: 2ra6. [Crossref]
- Biet M, Morin N, Lessard-Beaudoin M, Graham R, Duss S, et al. (2015) Prolongation of Action Potential Duration and QT Interval During Epilepsy Linked to Increased Contribution of Neuronal Sodium Channels to Cardiac Late Na+ Current Potential Mechanism for Sudden Death in Epilepsy. Circ Arrhythm Electrophysiol 8: 912-920. [Crossref]
- Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, et al. (2013) Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 123: 1798-1808. [Crossref]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, et al. (2006) Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116: 3127-3138. [Crossref]
- Erickson JR (2014) Mechanisms of CaMKII Activation in the Heart. Front Pharmacol 5: 59. [Crossref]
- Erickson JR, He BJ, Grumbach IM, Anderson ME (2011) CaMKII in the cardiovascular system: sensing redox states. Physiol Rev 91: 889-915. [Crossref]
- Luo M, Anderson ME (2013) Mechanisms of altered Ca2+ handling in heart failure. Circ Res 113: 690-708. [Crossref]
- Lu C-J, Hao G, Nikiforova N, Larsen H, Liu K, et al. (2015) CAPON Modulates Neuronal Calcium Handling and Cardiac Sympathetic Neurotransmission During Dysautonomia in Hypertension. Hypertension. 65:1288-1297. [Crossref]
- Chang K, Barth A, Sasano T, Kizana E, Kashiwakura Y, et al. (2008) CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci USA. 105: 4477-82. [Crossref]
- Kao W, Arking D, Post W, Rea T, Sotoodehnia N, et al. (2009) Genetic Variations in NOS1AP are Associated with Sudden Cardiac Death in U.S. White Community Based Populations. Circulation. 119: 940-951. [Crossref]
- Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, et al. (2009) NOS1AP is a genetic modifier of the long-QT syndrome. Circulation 120: 1657-1663. [Crossref]
- Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, et al. (2010) Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol 55: 2745-2752. [Crossref]
- Englot D, Konrad P, Morgan V (2016) Regional and global connectivity disturbances in focal epilepsy, related neurocognitive sequelae, and potential mechanistic underpinnings. Epilepsia 57: 1546-1557. [Crossref]
- Haneef Z, Chiang S, Yeh HJ, Engel J Jr, Stern JM (2015) Functional connectivity homogeneity correlates with duration of temporal lobe epilepsy. Epilepsy Behav 46: 227-233. [Crossref]