Abstract
Diabetes mellitus (DM) is a global metabolic disease with prevalence rates which have reached pandemic levels. This disease is strongly associated with vascular complications, even when the hyperglycemia is kept under control. The main complications of DM are stroke, myocardial infarction, and peripheral artery disease, and they often increase the risk of developing atherosclerosis, inflammatory angiopathy, and thrombosis. Angiopathy is commonly initiated by microvascular inflammation characterized by neutrophil extracellular traps (NETs) and metabolic abnormalities, and is controlled by (epi)-genetic events and immune / antigen-presenting cells. NETs play an important role in blood coagulation, activation of the innate and adaptive immune system, as well as vascular integrity and endothelial dysfunction. The aim of the review is to summarize the current knowledge about the role of NETs in the pathogenesis of vascular complications in DM.
Key words
diabetes mellitus; vascular complications; inflammation; atherosclerosis; neutrophil extracellular traps.
Introduction
Diabetes mellitus (DM) is one of the most important causes of premature death, disability and vascular complications worldwide [1]. The prevalence of DM steadily rises in developing and developed countries and has been achieved epidemic level. On the one hand, the prevalence of vascular events (stroke, myocardial infarction, peripheral artery disease) in diabetics frequently corresponds to occurrence of cardiovascular (CV) risk factors, such as hypertension, overweight / obesity, smoking, and dyslipidemia. On the other hand, developing DM strongly associates with endothelial dysfunction, microvascular inflammation and accelerating atherosclerosis, which negatively impact on short-term and long-term prognosis and relate to worsening quality-of-life [2,3]. However, DM-induced angiopathy predominantly accompany with type 1 DM (T1DM) and type 2 DM (T2DM) and does not closely relate to chronic hyperglycemia, which represented by glycated hemoglobin and glycemic variability [4]. Developing angiopathy in DM associates with early accelerating of atherosclerosis, stroke, unstable angina / myocardial infarction, blinding, advanced limb ischemia, nephropathy, and thrombotic complications [5-7]. Moreover, the risk of death due to vascular causes dramatically rises up to eight fold in DM patients with angiopathy in comparison with those who had no this complication [8]. Additionally, angiopathy due to other metabolic and inflammatory causes exhibited a positive liner correlation to a risk of death due to CV disease [6,7].
In this context, the common factor that directly triggers angiopathy is microvascular inflammation, which corresponds to metabolic abnormalities and is under tight control of (epi)-genetic regulation and immune / antigen-presenting cells [9-11]. Indeed, hyperglycemia alone, metabolic abnormalities, oxidative stress components (reactive oxygen species, advanced glycation end products), chemokines, cellular adhesive molecule, hormones (endothelin-1, aldosterone, angiotensin-II) interact with neutrophils and macrophages via several intercellular signaling pathways (i.e. PI3K/Akt / eNOs / NF-κB and ERK1/2 / p38 MAPK-activated protein kinases) and recruit predominantly neutrophil subsets in the vasculature promoting inflammation through not just synthesis and releasing of pro-inflammatory cytokines (tumor necrosis factor-alpha, interleukin-[IL]-2, IL-8, adiponectin, vistafin), but shaping of neutrophil extracellular traps (NETs) [12].
Recently NETs are considered as potent anti-microbial, anti-fungi, anti-viruses and anti-parasitic mechanism occurs with releasing of decondensated chromatin with a wide range of granular and intracellular proteins from some populations of activated neutrophils [13]. Therefore, NETosis plays an important role in blood coagulation, activation of innate and adaptive immune system, as well as vascular integrity and endothelial dysfunction [14,15]. Previous studies yielded that NETs appeared to be a link between endothelium, inflammation and thrombosis and that NETosis is crucial for development of vasculopathy due to numerous causes including metabolic diseases [16, 17]. The aim of the review is to summarize current knowledge regarding the role of NETs in pathogenesis of vascular complications in DM.
Definition of neutrophil extracellular trap
The NETs or NETosis is unique form of cell death, which is a core component of innate immune system that has been actively investigated for the last two decades [13]. NETosis is web-like structures composed of nuclear material and neutrophil granular proteins, which directly relate to releasing of modified chromatin in extracellular space due to several factors, i.e. pathogens, lipopolysaccharide, thromboxane A2, β-defensin-1, P-selectin/ P-selectin glycoprotein ligand-1, activated platelets, metabolic triggers (peptidyl-arginine deiminase 4 and nuclear DNA-binding protein of the high-mobility group box 1 - HMGB1), oxidative stress components (free radicals, oxygen ions, superoxide, hydrogen peroxide, and hypochloride), and even simple contact of various cells with NETosis-releasing cells [18].
Molecular mechanisms of neutrophil extracellular trap
Although extracellular factors are essential for initiating of NETosis, it is suggested that this process is under direct regulation of phagocyte Nox2 and it can be related to either redox imbalance or receptor-depending pathway [19]. Consequently, there are two major types of canonical NETosis, which distinguish one another with mechanisms regulating cell death (Figure 1). Classical canonical NETosis is mediated by NADPH oxidase 2 (NOX2), which is a highly regulated membrane-associated multiple protein complex that produce reactive oxygen species (ROS) including superoxides, hypochlorite, and peroxides leading to an oxidative stress, mitochondrial dysfunction, massive nuclear vacuolization and eventually intracellular chromatin decondensation and plasma membrane disintegration. Recent studies have shown that peptidyl arginine deiminase 4 (PAD4), elastase and myeloperoxidase (MPO) play a pivotal role in classical NETosis acting as mutually interplayed triggers [20-22]. For instance, PAD4 is able to catalyze histone citrullination and thereby induces chromatin decondensation, which may also be activated by MPO and elastase. After initiation of NETosis, elastase migrates to the nucleus and destroy histones and then this process may be supported by MPO [23].
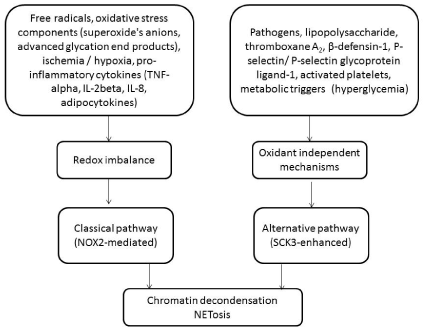
Figure 1: Diagram illustrating the two main pathways of NETosis.
Notes: NOX2, NADPH oxidase 2; IL, interleukin; TNF, tumor necrosis factor; SCK3- small conductance potassium channel-3.
Additionally, it was confirmed with the results of studies regarding pharmacological suppression of phorbol myristate acetate (PMA)-induced NADPH oxidase activity that prevents intracellular chromatin decondensation and consequently NET formation [24]. On the other hand, autophagy is not fully controlled by pharmacological inhibition of NADPH oxidase and tumor necrosis factor-alpha (TNF-alpha) and the fact has emphasized the apoptotsis could be a sort of back-up program for NETosis [24]. In this way, mitochondrial-induced ROS generation is probably not initial trigger of intracellular chromatin decondensation.
Alternative type of NETosis is called NOX-independent NETosis and it closely associates with opening of a voltage-dependent calcium-activated small conductance potassium (SCK) channel member SCK3 that directly control apoptosis [25]. It is confirmed the fact that neutrophils can produce NETs without previous activation of the NADPH oxidase complex with tight relation to several extracellular stimuli (growth factors, cytokines, mitogens, hormones, oxidative stress components, heat shock proteins). Perhaps, MPO and neutrophil elastase play co-regulating role in this process acting as triggers of delivery of ROS into mitochondria and thereby mediating mitochondrial dysfunction, accelerating ROS shaping, inducing cell death and NET formation [26]. Additionally, MPO may directly lead to dissociation of DNA from histones and thereby attenuate NETosis [27].
Although the activation of Akt and p38 mitogen-activated protein kinases (MAPK)-mediated signaling pathway is essential in both pathways, non-classical NETosis in contrast to classical cellular redox imbalance-induced NETosis can be activated through oxidant-independent mechanisms [28] and realize through Raf-MEK-extracellular signal regulated kinase 1/2 [29]. Finally, chromatin decondensation, which is core element of NETosis, dependent upon the activity of NADPH oxidase in classic pathway, otherwise neutrophil elastase and MPO is crucial in alternate pathway of NETosis [30,31]. However, both NOX-related and MPO-derived ROS production, but not mitochondrial ROS shaping are extremely important for and down-regulation of anti-apoptotic proteins and the release of NETosis [32]. Nevertheless, unlike PMA calcium- and protein arginine deiminase 4-dependent NETosis is initiated without previous neutrophil activation and degranulation. Additionally, there is sufficient difference between both types of NETosis in involvement of several molecular mechanisms activating neutrophil cell surface receptors. In fact, canonic Ras/Raf-1/MAPK/ERK kinase (Mek)/extracellular signal-regulated kinase (Erk) pathway is essential in spontaneous NETosis, while PI3K-dependent, non-canonical, pro-apoptotic FcγR-PI3Kβ/δ-Cdc42-Pak-Mek-Erk signaling pathway may play a pivotal role in immune-complex-driven human neutrophil activation [33, 34]. All these facts confirm that NETosis is potentially regulated receptor-mediating process. Indeed, prostaglandin E2, protein C can inhibit NETosis via its cognate receptor or through cooperation with protease-activated receptor 3 (PAR3) and CD11b/CD18 (Mac-1) integrins. In contrast, other factors (hypoxia, ischemia, bicarbonate levels) may activate NETosis, while there is not strong evidence regarding the role of hypoxia-inducing factor 1-alpha in this way. Probably, hypoxia / ischemia are able to increase the content of membrane-associated cholesterol, which could affect cell signaling loci, lipid micro domains, membrane rigidity and macrophage activation [35]. Thus, canonical ERK-1/2 signaling pathway was found as trigger of both responses, e.g. attenuation of cell survival by the post-translational modification of pro-survival genes as well as inactivation of a component of the apoptosis. On the other hand, none-canonical ERK-1/2 signaling pathway was rather apoptosis-enhanced machinery that pro-survival mechanism.
Finally, regardless of initial stimuli and pathways decondensed nuclear chromatin mixes other cytoplasmic components and granular membranes of cells, which is appeared due to massive vacuolization, and turn into extracellular space and eventually shapes NETs. NETs contain proteases (proteinase-3, neutrophil elastase), oxidases (MPO, NADPH oxidase, superoxide dismutase), wide range of bactericidal peptides, several post-translational modifications of histone / DNAs and ribonucleoproteins, which play a pivotal role in the trap and killing of hosts, but an exaggerated NETosis may directly lead to tissue damage [36]. Interestingly, that prior to plasma membrane rupture and NETosis the generation of many vesicles in neutrophils was found [37]. These vesicles have a double phospholipid bilayer and are believed to originate from the nuclear envelope [38], which disintegrates during NET cell death.
NETs have been implicated in numerous infections, trauma, sepsis, eclampsia, inflammation, thrombosis, autoimmune reactions, and malignancy [39]. In fact, chromatin modifications and NETs formation is mutually co-regulating processes that are coordinated by means gene transcription and RNA polymerase II elongation [40]. Consequently, deiminated chromatin is internalized on surface of the endothelium and mediates its biological effect through externalization of modified autoantigens, producing of type I interferon, stimulating the inflammasome shaping, and activating both the classic and alternative complement pathways [41,42]. Finally, NETs influence negatively on an ability of progenitor endothelial cells to proliferation, migration, differentiation and thereby it plays a pivotal role in impairment of vascular repair accompanying endothelial dysfunction, atherosclerotic plaque accumulation, and thrombosis [43]. Thus, NETosis is typical immune process that protects against invasion of hosts, but also it able to coordinate immune response with an activation of endothelial cells, coagulation, thrombosis, and microvascular inflammation.
NETosis in diabetes mellitus
Although the molecular basis responsible for angiopathy and atherosclerosis in DM has been deeply investigated, the innate mechanisms of the disease remain not fully clear [9,10]. It was well established that angiopathy in DM is result in various processes, such as exaggerated oxidative stress, impaired vascular repair, acceleration of atherosclerosis, altered endothelial function, systemic and microvascular inflammation due to fluctuated hyperglycemia and remarkable lipotoxicity [10]. Previous studies have shown that the neutrophils received from peripheral blood of diabetics exhibited spontaneous NETosis and that NETosis is enriched several components, such as elastase, histones, NADPH oxidase and protein kinase C [44]. Hyperglycemia demonstrated an ability to induce NETosis and associated with increased levels of circulating markers of NETosis including free cell DNA, elastase, mono- and oligonucleosomes [45]. In fact, though basal levels of NETs in patients with established T2DM were higher compared to healthy volunteers and related to fasting hyperglycemia, an attenuation of glycemia status with metformin was not completely associated with suppressed activity of TNF-alpha- and PMA-induced NETosis [46]. Indeed, Carestia A, et al. [46] reported that hyperglycemia was controlled in 6 months of metformin treatment, although a basal level and stimulated form of NETosis exhibited normal values in 12 months together with circulating biomarkers of NETosis, such as IL-6, TNF-alpha, cell-free DNAs. Joshi MB, et al. [47] have yielded a results regarding a formation of NETosis in neutrophils exposed to high glucose level as well as homocysteine and, IL-6. Authors believed that hyperglycemic conditions closely related to homocysteine, but homocysteine led to accelerated NETosis via both NADPH oxidase dependent and independent mechanisms in glucose interdependent manner. Thus, it has been suggested that increased level of NETosis in patients with T2DM did not occur to an impaired glycemic control but rather related to inflammation. In this context, spontaneous and inducible NETosis in T2DM may relate to different molecular pathways.
NETosis in vascular complications
It is well known that activated macrophages plays a pivotal role in pathogenesis of atherosclerosis supporting inflammatory reaction in plaque and leading to plaque instability [17]. Previous studies have now well established that activated macrophages are able to induce NETosis and that this mechanisms can tie atherosclerosis and intra plaque thrombosis [17,48]. Additionally, numerous non-specific stimuli, i.e. ischemia / hypoxia, oxidized lipoproteins, free fatty acids, necrotic cells, and modified histones, may be triggers for neutrophils to release NETs via represent an endogenous danger signal, which are able to activate stress-responsive transcription factor Nrf2 and regulate synthesis of inflammasome with pro-inflammatory cytokines (IL-1 alpha and IL-1 beta) [49-51]. Additionally, NETs are essential components of plaques and probably they contribute to generation of autoantibodies that lead to an aggravation of inflammation-related injury of vasculature. There is evidence that atherosclerosis may be accelerated by augmenting NETs formation within the plaque beyond autoantibodies’ shaping throughout activation of peripheral blood polymorphonuclear myeloid-derived suppressor cells, but not of inflammatory monocytes / macrophages [51,52]. Interestingly, Pertiwi KR et al. [53] have found that neutrophils and NETs were more frequently present in non-organized acute thrombi. Authors concluded that NETosis could be a prominent pro-thrombotic player in all distinct types of atherosclerosis and thrombosis, which facilitate the progression of vascular complications [53] and thus the onset of ensuing clinical peripheral artery ischemic syndromes. Thus, the molecular mechanisms of cell programmed death leading to apoptosis and also NETosis link inflammation and atherosclerosis, contribute to accumulation of pro-atherogenic, thrombotic factors and cell debris in circulation and thereby mediate progression of the peripheral artery disease (PAD).
Clinical studies have shown that elevated levels of serum dsDNA as a marker of NETosis was correlated with the presence of CV disease, atherosclerosis, nephropathy, and PAD [54,55]. There is evidence that angiopathy that appeared prior to atherosclerotic plaque accumulation and associated with endothelial dysfunction was closely linked to apoptotic endothelial cell originated chromatin-contained micro vesicles, which are trigger of NETosis [56,57]. Furthermore, cell free dsDNA levels associated positively with morphological evidence of plaque destabilization, severity of PAD and the risk of limb ischemia, as well as CV mortality rate [57,58]. Additionally, cell free DNA has not just cytotoxic effect, but can be a trigger of pro-thrombotic responses [59]. Probably, spontaneous / inducible NETosis could be a biomarker of higher risk of microvascular inflammation, endothelial dysfunction, thrombotic complication and atherosclerosis [60].
Previous studies have shown that activated neutrophils can promote hypercoagulation with NETosis and that they may be an active part of the thrombus formation [61,62]. Indeed, activated complement proteins activate NETs formation and interact with inflammation and coagulation [63]. Interestingly, an impact of activated macrophages and bone marrow-derived cells on vascular complications in CV disease seemed to be reported controversially in several studies. For instance, PAD4 produced by bone marrow-derived neutrophils and previously reported as a main trigger of NETs did not promote plaque in chronic experimental model of atherosclerosis, whereas it remained a leading cause in acute thrombus shaping in intimal lesions [63]. Authors concluded that the thrombotic complications in the plaque could relate to NETosis, but atherosclerosis did probably not [63]. On the other hand, cell-death-associated nuclear factors, such as DNAs, histones and nucleosomes, are actively released into the extracellular space by neutrophils through NETosis and directly contribute to factor VII activating protease acting as pro-coagulants [64]. On the other hand, cell-death-associated nuclear factors predominantly histones and cell-free DNAs were involved indirectly in coagulation cascade after releasing of factor XII from injured vascular wall [65]. Finally, factor VII activating protease together activated protein C is able to suppress histone cytotoxicity and thereby neutralizes or diminishes damaging effect of NETosis on vasculature and prevents coagulation [66,67]. Nevertheless, these effects were supported by hemodynamic forces, such as shear stress, they were not abrogated by inhibitors of PAD4, cyclooxygenase, platelet-neutrophil adhesion, high-mobility group protein box 1-receptor for advanced glycation end products interaction, and ATP/ADP [68,69]. Thus, NETosis may lead to pro-thrombotic state, while relationship between an ability of neutrophils to shape NETs-related pro-coagulation state depending on metabolic co-morbities including T2DM requires to be investigated in detail in the future.
In conclusion, in patients with DM vascular injury, endothelial dysfunction, accelerating atherosclerosis could be induced by metabolic signals that corresponding to both spontaneous and inducible NETosis for neutrophils. Probably, neutrophils could be a target for personified medical care in diabetics with higher risk of NETs-related vascular complications. However, the predictive value of NETs-related organ damages in DM requires to be investigated in large clinical trials in the future.
Funding and grants
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
conflict of interests
Not declared
References
- Mainous AG, Tanner RJ, Jo A, Park K, De Rochars VMB (2018) Trends in Cardiovascular Disease Risk in the U.S., 1999-2014. Am J Prev Med.
- Schuett KA, Lehrke M, Marx N, Burgmaier M (2015) High-Risk Cardiovascular Patients: Clinical Features, Comorbidities, and Interconnecting Mechanisms. Front Immunol 6: 591. [Crossref]
- Tong L, Chi C, Zhang Z (2018) Association of various glycemic variability indices and vascular outcomes in type-2 diabetes patients: A retrospective study. Medicine 97: e10860.
- Caprnda M, Mesarosova D, Ortega PF, Krahulec B, Egom E, et al. (2017) Glycemic Variability and Vascular Complications in Patients with Type 2 Diabetes Mellitus. Folia Med (Plovdiv) 59: 270-278. [Crossref]
- Wei F, Sun X, Zhao Y, Zhang H, Diao Y, et al. (2016) Excessive visit-to-visit glycemic variability independently deteriorates the progression of endothelial and renal dysfunction in patients with type 2 diabetes mellitus. BMC Nephrol 17: 67. [Crossref]
- Kakuta K, Dohi K, Miyoshi M, Yamanaka T (2017) Impact of renal function on the underlying pathophysiology of coronary plaque composition in patients with type 2 diabetes mellitus. Cardiovasc Diabetol 16: 131. [Crossref]
- Health Quality Ontario (2010) Stenting for peripheral artery disease of the lower extremities: an evidence-based analysis. Ont Health Technol Assess Ser 10: 1-88. [Crossref]
- Mohammedi K, Woodward M, Hirakawa Y, Zoungas S, Colagiuri S, et al (2016) ADVANCE Collaborative Group. Presentations of major peripheral arterial disease and risk of major outcomes in patients with type 2 diabetes: results from the ADVANCE-ON study. Cardiovasc Diabetol 15: 120-129.
- Marone EM, Cozzolino P, Ciampichini R, Chiodini V, Ferraresi R, Rinaldi LF, et al (2018) Peripheral Arterial Disease in diabetic patients: a long-term population-based study on occurrence, outcomes and cost. J Cardiovasc Surg 1: 2-5.
- Lehoux S, Jones EA (2016) Shear stress, arterial identity and atherosclerosis. Thromb Haemost 115: 467-473. [Crossref]
- Yamagishi S, Nakamura N, Suematsu M, Kaseda K, et al. (2015) Advanced Glycation End Products: A Molecular Target for Vascular Complications in Diabetes. Mol Med 21 Suppl 1: S32-40. [Crossref]
- Leavy O (2015) Inflammation: NETting a one-two punch. Nat Rev Immunol 15: 526-527. [Crossref]
- Kobayashi Y (2015) Neutrophil biology: an update. EXCLI J 14: 220-227. [Crossref]
- Yipp BG, Kubes P (2013) NETosis: how vital is it? Blood 122: 2784-2794. [Crossref]
- Martinod K, Wagner DD (2014) Thrombosis: tangled up in NETs. Blood 123: 2768-2776. [Crossref]
- Wong SL, Demers M, Martinod K, Gallant M, Wang Y, et al. (2015) Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 21: 815-819. [Crossref]
- Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, et al. (2015) NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol 52: 497-503. [Crossref]
- Leshner M, Wang S, Lewis C, Zheng H, Chen XA, et al. (2012) PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 3: 307. [Crossref]
- Farrera C, Fadeel B (2013) Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 191:2647–2656
- Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, et al. (2011) Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117: 953-959. [Crossref]
- Björnsdottir H, Welin A, Michaëlsson E, Osla V, Berg S, et al. (2015) Neutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic Biol Med 89: 1024-1035. [Crossref]
- Palladino END, Katunga LA, Kolar GR (2018) 2-Chlorofatty acids: lipid mediators of neutrophil extracellular trap formation. J Lipid Res 59: 1424-1432. [Crossref]
- Leshner M, Wang S, Lewis C, Zheng H, Chen XA, et al. (2012) PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 3: 307. [Crossref]
- Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, et al. (2011) Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21: 290-304. [Crossref]
- Fay AJ, Qian X, Jan YN, Jan LY (2006) SK channels mediate NADPH oxidase-independent reactive oxygen species production and apoptosis in granulocytes. Proc Natl Acad Sci U S A 103: 17548-17553. [Crossref]
- Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. Journal of Cell Biology 191: 677-691
- Palladino END, Katunga LA, Kolar GR (2018) 2-Chlorofatty acids: lipid mediators of neutrophil extracellular trap formation. J Lipid Res 59: 1424-1432. [Crossref]
- Douda DN, Khan MA, Grasemann H, Palaniyar N (2015) SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A 112: 2817-2822. [Crossref]
- Matsubayashi Y, Fukuda M, Nishida E (2001) Evidence for existence of a nuclear pore complex-mediated, cytosol-independent pathway of nuclear translocation of ERK MAP kinase in permeabilized cells. The Journal of biological chemistry 276: 41755-41760
- Rochael NC, Guimarães-Costa AB, Nascimento MT, DeSouza-Vieira T (2015) Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci Rep 5: 18302. [Crossref]
- Nishinaka Y, Arai T, Adachi S, Takaori-Kondo A, Yamashita K (2011) Singlet oxygen is essential for neutrophil extracellular trap formation. Biochemical and Biophysical Research Communications 413: 75-79
- Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, et al. (2011) Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7: 75-77. [Crossref]
- Chu JY, Dransfield I, Rossi AG, Vermeren S (2016) Non-canonical PI3K-Cdc42-Pak-Mek-Erk Signaling Promotes Immune-Complex-Induced Apoptosis in Human Neutrophils. Cell Rep 17: 374-386. [Crossref]
- Douda DN, Yip L, Khan MA, Grasemann H, Palaniyar N (2014) Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood 123: 597-600. [Crossref]
- Kaplan MJ, Radic M (2012) Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189: 2689-2695. [Crossref]
- Neeli I, Khan SN, Radic M (2008) Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 180: 1895-1902. [Crossref]
- Wang H, Wang Q, Venugopal J, Wang J, Kleiman K, et al. (2018) Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci Rep 8: 4881. [Crossref]
- Qi H, Yang S, Zhang L (2017) Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front Immunol 8: 928. [Crossref]
- Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC (2012) Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol 92: 841-849. [Crossref]
- Nakashima K, Hagiwara T, Yamada M (2002) Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem 277: 49562-49568. [Crossref]
- Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, et al. (2015) PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 162: 1003-1015.
- Wang H (2015) Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol 181: 518-527. [Crossref]
- Ma YH (2016) High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res Ther 18: 2. [Crossref]
- Berezin AE (2016) The Neutrophil Extracellular Traps: The Missed Link between Microvascular Inflammation and Diabetes? Metabolomics. 6: 163-166.
- Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, et al. (2016) NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 65: 1061-1071. [Crossref]
- Carestia A, Frechtel G, Cerrone G, Linari MA, Gonzalez CD, et al. (2016) NETosis before and after Hyperglycemic Control in Type 2 Diabetes Mellitus Patients. PLoS One 11: e0168647. [Crossref]
- Joshi MB, Baipadithaya G, Balakrishnan A, Hegde M, Vohra M, et al. (2016) Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci Rep 6: 36362. [Crossref]
- Sollberger G, Tilley DO, Zychlinsky A (2018) Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev Cell 44: 542-553. [Crossref]
- Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, et al. (2015) NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol 52: 497-503. [Crossref]
- Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V (2015) Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349: 316-320. [Crossref]
- Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, et al. (2013) Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol 14: 1045-1053. [Crossref]
- Yamamoto K, Yamada H, Wakana N, Kikai M, Terada K, Wada N, et al (2018) Augmented neutrophil extracellular traps formation promotes atherosclerosis development in socially defeated apoE-/- mice. Biochem Biophys Res Commun 500: 490-496.
- Pertiwi KR, van der Wal AC, Pabittei DR, Mackaaij C (2018) Neutrophil Extracellular Traps Participate in All Different Types of Thrombotic and Haemorrhagic Complications of Coronary Atherosclerosis. Thromb Haemost 118: 1078-1087. [Crossref]
- Fadini GP, Menegazzo L, Scattolini V, Gintoli M, Albiero M, et al. (2016) A perspective on NETosis in diabetes and cardiometabolic disorders. Nutr Metab Cardiovasc Dis 26: 1-8. [Crossref]
- Mangold A, Alias S, Scherz T, Hofbauer T, Jakowitsch J, et al. (2015) Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res 116: 1182-1192. [Crossref]
- Berezin AE (2016) “Impaired immune phenotype" of endothelial cell-derived microparticles: the missed link between diabetes-related states and cardiovascular complications? Journal of Data Mining in Genomics & Proteomics 7: 195-197
- Berezin AE (2016) Is the neutrophil extracellular trap-driven microvascular inflammation essential for diabetes vasculopathy? Biomedical Research and Therapy 3: 618-624.
- Berezin AE (2016) The cell-free mitochondrial DNA: a novel biomarker of cardiovascular risk? Translational Biomedicine 7: 68-71
- Mozzini C, Garbin U, Fratta Pasini AM, Cominacini L (2017) An exploratory look at NETosis in atherosclerosis. Intern Emerg Med 12: 13-22. [Crossref]
- Megens RT, Vijayan S, Lievens D, Doring Y, van Zandvoort MA, et al (2012) Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost. 107: 597-598
- Morel O, Jesel L, Abbas M, Morel N (2013) Prothrombotic changes in diabetes mellitus. Semin Thromb Hemost 39: 477-488. [Crossref]
- Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, et al. (2013) Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 110: 8674-8679. [Crossref]
- de Bont CM, Boelens WC, Pruijn GJM (2018) NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol 5: 5-10. [Crossref]
- Marsman G, von Richthofen H, Bulder I, Lupu F, Hazelzet J, et al. (2017) DNA and factor VII-activating protease protect against the cytotoxicity of histones. Blood Adv 1: 2491-2502.
- Elaskalani O, Abdol Razak NB, Metharom P (2018) Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal 16: 24. [Crossref]
- Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM, Key NS (2017) In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 129: 1021-1029.
- Stephan F, Dienava-Verdoold I, Bulder I, Wouters D, Mast AE, et al. (2012) Tissue factor pathway inhibitor is an inhibitor of factor VII-activating protease. J Thromb Haemost 10: 1165-1171. [Crossref]
- Yu X, Tan J, Diamond SL (2018) Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J Thromb Haemost 16: 316-329. [Crossref]
- Martinod K, Witsch T, Farley K, Gallant M, Remold-O'Donnell E, et al. (2016) Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost 14: 551-558.