In recent years, various clinical trials are exploring the efficacy of cell-based strategies to treat cartilage defects. Human adipose-derived stem/stromal cells (hASCs) have great potential for the regeneration of articular cartilage due to their ability to undergo differentiation into the chondrogenic lineage. In most cell-based therapies, the single dose requires 107-109 cells, and then it is necessary to move from manual flask-based culture to bioreactor whose monitoring and control of biological variability is necessary. Furthermore, before clinical use, the success of hASC differentiation need to be thoroughly inspected. In this study, we sought to assess with complementary methods the chondrogenic potential of hASCs expanded in flask or in a hollow-fiber bioreactor (HFBR). Human ASCs, after phenotypic characterization were subjected to chondrogenic differentiation, the expression of cartilage-specific genes was analysed by quantitative real-time polymerase chain reaction (qPCR); optical microscopy as well as transmission electron microscopy (TEM) were also performed. The two culture expansion systems do not affect hASC immunophenotype; microscopy analysis confirmed the successful of hASC differentiation process. Gene expression analysis after differentiation displayed an upregulation of cartilage-specific genes more noticeable in hASCs expanded in the HFBR, suggesting that hASCs expanded in the HFBR can be efficiently differentiated into chondroblasts. Our study demonstrates that flask and HFBR expansion methods do not affect the subsequent differentiation process; additionally, our results highlight the importance of analyzing differentiation by different techniques and support the importance of TEM analysis that is able to give additional critical information to molecular biology results.
collagen fibers, chondrogenesis, hASCs, histochemistry, qPCR, TEM
Articular hyaline cartilage has limited regenerative ability, and the loss of its function can frequently be painful and debilitating. Thus, efforts have been made to intervene in cartilage defects with a regenerative medicine approach aiming at supporting biological repair with cell-based strategies for in situ regeneration of the tissue [1]. In this perspective, mesenchymal stem cells (MSCs) represent an appealing cell source for cartilage repair.
MSCs are multipotent cells with self-renewal ability [2,3] and capable of differentiating into various cell types [4-8]. MSCs are also categorized as stromal cells with tissue regenerative and remodeling abilities due to the actions exerted by the release of bioactive molecules with trophic, immunomodulatory, anti-scarring and chemo-attractant activities [9-12]. MSCs can be obtained from several tissues, bone marrow, adipose tissue, peripheral blood, placenta, and umbilical cord [13]. In recent years, adipose tissue has gained increased interest as a source of MSCs because it is more easily accessible and can be obtained in large quantities by liposuction under local anesthesia with minimum discomfort for patients [14].
hASCs are found in the stromal vascular fraction (SVF) of adipose tissue and seem to be as effective as bone marrow-derived MSCs in clinical application, and, in some cases, may be better suited [15-18]. Although the number of cells depends on therapeutic applications, in most MSC-based therapies the single dose requires 107-109 cells. It is clear that it is necessary to move cell expansion from manual flask-based culture to bioreactor. Despite the high level of automation and precision, the monitoring and the control of biological variability of the bioreactor becomes important [19].
The pellet culture method is a widely applied system for the induction of in vitro chondrogenesis of MSCs [20]. In fact, cell-cell contacts seem to be a crucial prerequisite for the onset of chondrogenic differentiation [21]. Pellets are submitted to chondrogenic induction with a basal medium containing growth factors such as TGFB1, 2, and 3 which promote chondrogenesis and lead to deposition of proteoglycans and collagen type II when used as single factors [22]. Other factors such as bone morphogenetic protein 6 [23] and prostaglandin E2 [24], can be used in combination with the inducer TGFB.
However, before moving towards transplantation into patients, the success of differentiation and the maintenance of the chondroblast/chondrocyte phenotype need to be exhaustively investigated by means of various and complementary methods. The assessment of chondrogenesis is usually performed with well-established histochemical methods, such as Alcian Blue and Safranin-O staining for proteoglycans, and Masson’s trichrome for collagen fibers. Although a qualitative analysis of the differentiation based on histochemistry is helpful, it is not sufficient for rigorous analysis.
Gene expression analysis by real-time polymerase chain reaction has been a key technology of the post-genome era and has become a routine and robust approach for measuring the expression of genes of interest, validating microarray experiments, and monitoring biomarkers [25]. Hence, many researchers corroborate histochemical analysis with the analysis for the expression of lineage-specific genes [23]. In spite of that, since mRNA molecules are the first intermediaries of a highly sophisticated cell mechanism that includes transcriptional, post-transcriptional, translational and post-translational regulation [26], gene expression alone is not informative enough to define the protein profile. To support this concept, some groups have published data demonstrating that mRNA level alone could not predict protein profile [27-30]. Furthermore, it is known that the morphology of cells with the same genome may be different depending on their protein expression that in turn influences morphological characteristics while cells with different transcriptomes may present similar phenotype [31]. This is why in some studies, e.g. cell differentiation, it is appropriate to combine different methods such as transcriptomic and proteomic analysis or, as in this work, transcriptomic and morphological analysis. The morphological and structural analysis aims at knowing the shape, the size, the distribution and the structural organization of the cells and their relationship with extracellular matrix (ECM) components. The main techniques used for the morphological analysis are based on the use of optical and transmission electron microscopy, a tool that can provide ultrastructural information [32]. In our study, we aimed at comparing the chondrogenic differentiation of hASCs expanded in a HFBR or flask by using various complementary approaches: gene expression analysis, optical microscopy and TEM. We confirmed the success of differentiation of hASCs into chondroblasts and their ability to synthesize tropocollagen molecules, which are eventually exported in the extracellular space where they form collagen fibers. Based on the weight of the information obtained by TEM and their correlation with the other methodologies used in this study, we propose HFBR as valid and saving time method to expand hASCs without affect chondrogenic differentiation.
Sampling of adipose tissue
Liposuction was performed during surgical aesthetic procedures. Three women in good health and negative for human immunodeficiency, hepatitis B and C virus, were included in this study. All donors signed an informed consent declaration. Liposuction procedure started with a pre-emptive analgesia: Calecoxib 400 mg per oral administration 1 hour before surgery. Before going to the operating room, an intravenous infusion with 100 ml of NaCl 0.9%, Ranetidin 50 mg, Ondansetron 4 mg, Desametason 8 mg, Cefazolin 2 g was administered, and a sedation was performed with Midazolam 1 mg bolus intravenous. Sedoanalgesia was performed with intravenous Sufentanil bolus (0.05 µg/kg) and Propofol continuous infusion. The access points of the cannula were infiltrated with a physiologic solution containing 0.1% lidocaine and 1:100,000 adrenalin. A negative pressure of 400 mm Hg was applied to the cannula connected to a 60 ml syringe for aspiration. Adipose tissue samples were collected, anonymized and sent to our authorized research biobank facility (approval CE 2961, Ethical Committee Canton Ticino, Switzerland). All protocols were reviewed and approved also by the “University of Insubria” ethic committee in accordance with the Declaration of Helsinki.
SVF isolation and cryopreservation
The isolation of the SVF was performed by a protocol developed in our laboratories (Patent PCT/EP2012/069261). Briefly, 150 ml of adipose tissue was washed twice with Dulbecco’s PBS (DPBS with Ca2+ and Mg2+, Gibco, Life Technologies, Oregon, USA) in a 100-ml syringe (BBraun Medical AG, Melsungen, Germany) and held vertically in a support stand for few minutes to spontaneously separate adipose tissue and hydrophilic fluids. Aqueous phase was discarded and adipose tissue was digested with Liberase MNP-S (Roche Applied Science, Basel, Switzerland) at a final concentration of 0.28 Wünsch U/ml diluted in DPBS (with Ca2+ and Mg2+) and incubated at 37°C for 45 minutes under agitation. Enzymatic reaction was stopped by DPBS (without Ca2+ and Mg2+, Gibco, Life Technologies, Oregon, USA) supplemented with 1% albumin (CSL Behring AG, Bern, Switzerland) and strongly agitated to separate the hydrophilic phase from the hydrophobic one. The lower layer, which contains the SVF cells, was carefully poured out into a conical 50 ml centrifuge tube (Falcon, Corning Science, México) and washed with 1% albumin solution to increase cell yield. Finally, after filtration through a 100 μm and a 40 μm sieve (BD Falcon, Basel, Switzerland), the SVF was centrifuged at 400 x g at room temperature (RT) for 5 minutes and cells were resuspended in 5% albumin solution (CSL Behring AG, Bern, Switzerland). The SVF was then analyzed for cell count and viability using an automated propidium iodide-based cell counting device (Nucleocounter NC-100, Chemometec A/S, Allerod, Denmark). For cryopreservation, SVF cells were centrifuged 5 minutes at 400 × g, resuspended in an ice-cold solution of 1% albumin solution, 5.5% ME2SO and 4.5% dextran-40 (Cryosure DEX-40, WAK-Chemie Medical GmbH, Steinbach, Germany) in MEM alpha (PAA Laboratories, Pasching, Austria) and transferred into a 2 ml cryovial (Nalgene, Thermo Fisher Scientific, Waltham, USA). Cells were frozen by means of a programmable freezer (Consartic GmbH, Schoellkrippen, Germany) under the following conditions: from 4°C to 0°C in 6 minutes, then hold for 15 minutes at 0°C. From 0°C to -°C in 9 minutes and then hold at -2°C for 2 minutes. From -2°C to -35°C in 25.5 minutes and finally from -35°C to -100°C in 13 minutes. Cryovials were then transferred into liquid nitrogen for long-term storage. For thawing, the cryovial was immersed in a 37°C water bath for 2-3 minutes. Immediately after being thawed, the cells were carefully aspirated, mixed with 9 volumes of pre-warmed cell culture medium in a conical 50 ml centrifuge tube (Falcon, Corning Science, México), centrifuged at 400 × g for 5 minutes and resuspended in cell culture medium.
Cell culture
For HFBR-based cell culture, the disposable HFBR (Cell Expansion Set, Terumo BCT, Lakewood, USA) was loaded on the Quantum Cell Expansion System (Terumo BCT, Lakewood, USA), primed with DPBS (without Ca2+ and Mg2+, Gibco, Life Technologies, Oregon, USA) and coated overnight with human fibronectin (Corning, New York, USA) to promote cell adhesion. After coating the bioreactor, any excess fibronectin was washed from the bioreactor and the cell culture medium was introduced into the set and oxygenated for a minimum of 30 minutes. Cell culture medium consisted of 5% pooled human platelet lysate (hPL, Stemulate, Cook Regentec, Indianapolis, USA) in Ham’s F12/IMDM (1:1) medium (Cell Culture Technologies, Gravesano, Switzerland) with addition of primocin (InvivoGen, San Diego, USA) and L-alanyl-L-glutamine (GlutaMax, Gibco, Life Technologies, Oregon, USA). The gas mixture used was 74% N2, 21% O2, 5% CO2. At this point, thawed SVF cells were loaded in the HFBR at a concentration of 9.6-20 × 106 hASCs/cm2 and were allowed to adhere for 24-48 hours, followed by a rapid washout to remove any non-adherent cells. Cells were grown for 6-13 days and the inlet rate for medium perfusion was adjusted according to manufacturer’s instructions and was based on lactate concentration that was determined daily with a colorimetric method (Lactate Rapid, Diaglobal, Berlin, Germany). When lactate concentration reached 6-8 mM for 1-2 days, hASCs were harvested using TrypLE Select (Gibco, Life Technologies, Oregon, USA).
For flask culture, cells were seeded in flask (Nunc, Thermo Fisher Scientific, Waltham, USA) at the same conditions as for HFBR (see above). Cell harvest was performed by washing with DPBS (without Ca2+ and Mg2+, Gibco, Life Technologies, Oregon, USA) and incubating with TrypLE Select for 10 minutes at 37°C. Cell counting and assessing of viability was performed with an automated propidium iodide-based cell counting device (Nucleocounter NC-100, ChemoMetec A/S, Denmark).
Immunophenotyping by Flow Cytometry
SVF characterization was performed as previously described [33]. Briefly, after SVF isolation, 5 × 105 cells were resuspended in flow cytometry buffer and stained with the following monoclonal antibodies: CD146-PE, CD34-APC-A750, CD45-KrO (Beckman Coulter, Nyon, Switzerland), Syto 40 (Life Technologies, Oregon, USA) to exclude cellular debris and 7-amino-actinomycin D (7-AAD, Beckman Coulter, Nyon, Switzerland) to assess cell viability. After 20 minutes of incubation, erythrocytes were lysed using Versalyse lysing solution (Beckman Coulter, Nyon, Switzerland) and, before acquisition, Flow-Count Fluorospheres (Beckman Coulter, Nyon, Switzerland) were added, to directly determine the absolute number of hASCs. hASCs were identified as the CD45 and CD146 negative and CD34 positive fraction [18,33].
For immunophenotypic characterization of pre-selected hASCs, 5 × 105 cells were resuspended in flow cytometry buffer and incubated with LIVE/DEAD fixable stain (Life Technologies, Oregon, USA) at RT in the dark for 30 minutes. Cells were then washed in flow cytometry buffer and stained for 30 minutes at RT with control antibodies IgG1-FITC (Beckman Coulter, Nyon, Switzerland), IgG3-PE (Beckman Coulter, Nyon, Switzerland), IgG1-PC5 (Beckman Coulter, Nyon, Switzerland), IgG1-PC7 (Beckman Coulter, Nyon, Switzerland), IgG1-APC (Beckman Coulter, Nyon, Switzerland), IgG1-APC-A750 (Beckman Coulter, Nyon, Switzerland), IgG1-KrO (Beckman Coulter, Nyon, Switzerland) or with CD73-FITC (BD Biosciences, Bedford, USA), CD31-FITC (BD Biosciences, Bedford, USA), CD105-PE (Beckman Coulter, Nyon, Switzerland), CD90-PC5 (Beckman Coulter, Nyon, Switzerland), CD13-PC7 (Beckman Coulter, Nyon, Switzerland), CD11b-APC (Beckman Coulter, Nyon, Switzerland), CD44-APC-A750 (Beckman Coulter, Nyon, Switzerland), CD45-KrO (Beckman Coulter, Nyon, Switzerland). Before measurement, cells were resuspended in IOTest 3 Fixative Solution (Beckman Coulter, Nyon, Switzerland). All flow cytometry analyses were performed using a Navios 3-lasers/10-channels flow cytometer (Beckman Coulter, Nyon, Switzerland), and data were analyzed with Kaluza software (Beckman Coulter, Nyon, Switzerland).
Chondrogenic differentiation
Chondrogenic differentiation was performed by a protocol developed in our laboratories (Patent PCT/EP2013/072738). Briefly, 5 × 105 hASCs at passage 1 or 2 were gently centrifuged in a 50 ml conical Tubespin bioreactors (TPP, Trasadingen, Switzerland) to form small pellets and cultured for 21 days in a DMEM high glucose medium (Gibco, Life Technologies, Oregon, USA) supplemented with primocin (InvivoGen, San Diego, USA), L-alanyl-L-glutamine (GlutaMax, Gibco, Life Technologies, Oregon, USA), ascorbic acid 2-phosphate (0.1 mM, Sigma Aldrich, St. Louis, USA), human transferrin (5 µg/ml), human insulin (5 µg/ml), selenium (5 ng/ml) (ITS Premix, Corning, New York, USA), human albumin (250 µg/ml, CSL Behring AG, Bern), basic fibroblast growth factor (bFGF, 10 ng/ml, Prospec, Ness Ziona, Israel), human transforming growth factor-beta 3 (TGFB3, 10 ng/ml, Prospec, Ness Ziona, Israel), human bone morphogenetic protein-6 (BMP6, 10 ng/ml, Prospec, Ness Ziona, Israel), sodium pyruvate (1 mM, Gibco, Life Technologies, Oregon, USA), prostaglandin E2 (PGE2, 10-7 M, Prospec, Ness Ziona, Israel). Control cells were incubated in 5% pooled human platelet lysate (hPL, Stemulate, Cook Regentec, Indianapolis, USA) in Ham’s F12/IMDM (1:1) medium (Cell Culture Technologies, Gravesano, Switzerland) with addition of primocin (InvivoGen, San Diego, USA) and L-alanyl-L-glutamine (GlutaMax, Gibco, Life Technologies, Oregon, USA).
RNA Isolation and Reverse Transcription
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer’s instruction. The concentration of extracted RNA from each sample was determined by spectrophotometric analysis with a Jenway Genova spectrophotometer (Bibby Scientific Limited, Staffordshire, United Kingdom). cDNA was synthesized using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, USA) with oligodT and random hexamer primers according to manufacturer’s instruction. After reverse transcription, all cDNA samples were diluted to a final concentration of 5 ng/µl and stored at -80°C until use.
Quantitative Real-Time Polymerase Chain Reaction
qPCR analysis was performed by SYBR Green technology. Reactions were set up for 10 ng of cDNA in a final volume of 20 µl in a 96-well plate (Bio-Rad, Hercules, USA) and processed in a CFX Connect Real Time PCR Detection System (Bio-Rad, Hercules, USA). The PCR master mix used contained 1× SsoAdvanced SYBR Green Supermix (Bio-Rad, Hercules, USA), 250 nM forward primer, 250 nM reverse primer, up to 20 µl with nuclease-free water. Primers were designed by primer-BLAST (NCBI, USA), within the sequences of a panel of genes (ACAN, SOX9, COMP, COL1A1, COL2A1, COL3A1), on an exon-exon junction in order to prevent genomic DNA amplification. To analyze the relative expression of different genes, three housekeeping genes were chosen (TBP, ACTB and YWHAZ) and the geometric mean of their Ct values was calculated [34].
A sample without cDNA was used to verify the absence of nucleic acid contaminations. A cDNA sample composed of a mix of various cell extracts was used as calibrator. Thermocycler program consisted of an initial hot start cycle at 95°C for 30 seconds followed by 45 amplification cycles resulting in a denaturation step at 95°C for 10 seconds and an annealing-extension phase at 60°C for 30 seconds. At the end of the analysis, the temperature was raised from 65°C to 95°C, at 0.5°C/5 seconds, for melt-curve evaluation. For all samples, reactions were performed in duplicate. The Ct values were recorded with a threshold of 3000 relative fluorescence units and the relative gene expression, expressed as 2-ΔΔCt. Results are expressed as mean ± SD. Primers used in this work are reported in Table 1.
Table 1. Primers used in this study.
Gene Name |
Sequence 5’-3’ |
Type |
Amplicon length (bp) |
Accession Number |
TBP |
F: TGCACAGGAGCCAAGAGTGAA |
housekeeping |
131 |
NM_001172085 |
R: CACATCACAGCTCCCCACCA |
ACTB |
F: GCACAGAGCCTCGCCTTT |
housekeeping |
69 |
NM_001101 |
R: TATCATCATCCATGGTGAGCTGG |
YWHAZ |
F: TGGCTCGAGAATACAGAGAG |
housekeeping |
99 |
NM_001135699 |
R: GTGAAGCATTGGGGATCAAG |
ACAN |
F: GTCGTGGTGAAAGGCATCGTGTTC |
target |
112 |
NM_001135.3 |
R: GCGTGGCAATGATGGCACTGTTCTG |
SOX9 |
F: TCTGGAGACTTCTGAACGAG |
target |
127 |
NM_000346.3 |
R: TTCTTCACCGACTTCCTCC |
COMP |
F: AGAAGTCCTATCGTTGGTTCC |
target |
104 |
NM_000095 |
R: CAAGACCACGTTGCTGTC |
COL1A1 |
F: CCAGAAGAACTGGTACATCAGCAA |
target |
70 |
NM_000088.3 |
R: CGCCATACTCGAACTGGAATC |
COL2A1 |
F: ACCTCAAGAAGGCCCTGCTCATC |
target |
110 |
NM_001844.4 |
R: CGGTATGTTTCGTGCAGCCATCC |
COL3A1 |
F: GGGAACATCCTCCTTCAACA |
target |
183 |
NM_000090.3 |
R: GCAGGGAACAACTTGATGGT |
Abbreviations: F,Forward Primer; R,Reverse Primer
Optical microscopy analysis
For optical analysis, pellets were washed twice with DPBS (without Ca2+ and Mg2+, Gibco, Life Technologies, Oregon, USA), fixed in Bouin’s solution (Bio-Optica, Milano, Italy) for 2 hours at RT, dehydrated with increasing scale of ethanol, treated with xylene and finally embedded in paraffin. Sections (5 µm) were cut, deparaffinized with xylene, rehydrated with decreasing scale of ethanol and washed in distilled water. The specimens were stained with Alcian blue solution (1% in 3% acetic acid, pH 2.5, Sigma Aldrich, St. Louis, USA), Safranin-O/Fast Green (Sigma Aldrich, St. Louis, USA) and Masson’s trichrome (Bio-Optica, Milano, Italy). Alcian blue was applied for 45 minutes at RT. For Safranin-O/Fast Green, specimens were stained with Hematoxylin (Bio-Optica, Milano, Italy) for 5 minutes, washed in running water for 5 minutes and distained in Acid EtOH. Pellets were stained with Fast Green solution for 5 minutes, rinsed with 1% acetic acid solution for 15 seconds and finally stained with 0.1% Safranin-O solution for 5 minutes. For Masson’s trichrome, specimens were stained in Weigert’s iron hematoxylin working solution for 10 minutes, in picric acid alcoholic solution for 4 minutes and washed in distilled water. Pounced acid fuchsine and phosphomolybdic acid solution were applied for 4 and 10 minutes respectively. Aniline blue solution was used for collagen fibers staining. Finally, sections were mounted using EUKITT (Bio-Optica, Milan, Italy). Observations were performed by a Zeiss Axiophot microscope (Carl Zeiss AG, Werk Göttingen, Germany) under bright light illumination and photographs were acquired by Discovery C30 camera (Tucsen Imaging Technology Co., Ltd. Fujian, China).
Transmission electron microscopy analysis
For TEM studies, pellets were washed with DPBS (without Ca2+ and Mg2+, Gibco, Life Technologies, Oregon, USA) and fixed in Karnovsky solution (4% formaldehyde, 2% glutaraldehyde, 0.1 M sodium cacodylate, pH 7.2) overnight at 4°C. Samples were washed three times in 0.1 M sodium cacodylate for 10 minutes and postfixed in dark for 1 hour with 1% osmium tetroxide in 0.1 M sodium cacodylate-buffer (pH 7.2) at RT. After dehydration with a series of increasing scale of alcohol, samples were embedded in an Epon-Araldite 812 1:1 mixture. Thin sections (90 nm), obtained with a Pabisch Top-Ultra ultramicrotome (Emme 3 S.r.l., Milan, Italy), were stained with uranyl acetate and lead citrate according to the standard methods and observed with a Morgagni electron microscope (Philips, Eindhowen, NL) operated at 80 KeV.
Immunophenotyping by flow cytometry
Immunophenotyping was performed according to the International Society for Cell Therapy (ISCT) standards [18,35] to confirm the MSC nature of the cells harvested after cell culture in flask or HFBR, i.e. before subjecting cells to chondrogenic differentiation. All hASC cultures, independently of the expansion method, where found negative (<5%) for the endothelial marker CD31, the hematopoietic antigen CD45, and the leukocyte-specific marker CD11b. Concomitantly, cells where positive (>95%) for the conventional mesenchymal surface antigens CD105, CD90 and CD73, as well as the stromal markers CD13 and CD44 (Figure 1). No significant differences were found between hASCs cultured in flask or HFBR, hence these results confirm the validity of both methods for hASC expansion.
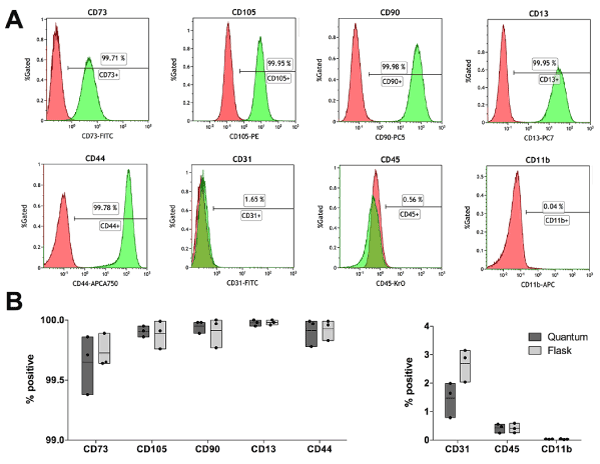
Figure 1. Typical flow cytometry analysis of hASCs after harvest from flask or HFBR. (A): Red histograms show the logarithmic fluorescence signal for the isotypic control stain (IgG1- FITC, IgG3-PE, IgG1-PC5, IgG1-PC7, IgG1-APC, IgG1-APC-A750, IgG1-KrO) whereas green histograms show the logarithmic fluorescence signal for the full stain (CD73-FITC, CD31-FITC, CD105-PE, CD90-PC5, CD13-PC7, CD11b-APC, CD44-APCA750, CD45-KrO). (B): Graphical representation of the level of positivity based on isotypic control for positive (CD73, CD90, CD105, CD13, CD44) and negative (CD31, CD11b, CD45) MSC markers. Single sample data (points) are shown in floating bars (representing the minimum to maximum range of distribution) with line at mean.
Expression of chondrogenesis-related genes
We analysed the expression of genes involved in chondrogenesis in control and induced hASCs after 21 days the start of the induction. The expression was considered up- or downregulated starting from a 2-fold difference between control and induced samples. Increased levels of chondrogenesis-related genes were observed in induced samples, particularly after HFBR expansion (Figure 2).
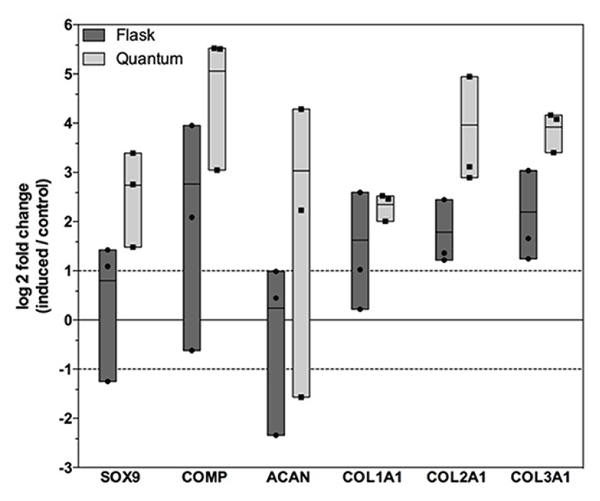
Figure 2. Expression of genes involved in hASC chondrogenic differentiation. Figure shows SOX9, ACAN, COMP, COL1A1, COL2A1 and COL3A1 in hASCs expanded in flask (light grey) and HFBR (dark grey) and submitted to chondrogenic differentiation. Shown are log 2 fold changes of induced samples compared to control (se to zero). Single sample data (points) are shown in floating bars (representing the minimum to maximum range of distribution) with line at mean. Dashed lines on the y-axis represent a 2-fold up- or down-regulation.
When compared to control samples, mRNA levels of SRY-box 9 (SOX9) [36], revealed a 1.74-fold higher expression in differentiated hASCs expanded in flask, while the expression was higher and could be considered up-regulated (6.68-fold) in those obtained from HFBR. In line with the increase of SOX9 transcripts, the expression of the cartilage-specific proteoglycan aggrecan (ACAN) [37] was not upregulated when flask-derived hASCs were used for differentiation (1.18-fold). On the contrary, chondrogenic induction of HFBR-derived cells revealed an 8.17-fold higher expression. The transcription of cartilage oligomeric matrix protein (COMP), encoding for a ECM glycoprotein that is abundant in tissues subjected to load [38], were up-regulated both for flask- (6.8-fold) and HFBR-derived (33.25-fold) hASCs.
The expressions of collagen type I alpha 1 (COL1A1), collagen type II alpha 1 (COL2A1) and collagen type III alpha 1 (COL3A1), the main ECM components in the cartilage [39], were upregulated for induced cells obtained from both systems (from 3.07 to 4.57-fold for the flask group, and from 5.08 to 15.62-fold for the HFBR group).
Optical microscopy analysis
The capability of hASCs, expanded with the two systems, to differentiate in chondroblasts was evaluated using Alcian Blue and Safranin-O, which stain proteoglycans and glycosaminoglycans of the ECM, and Masson’s trichrome, which distinguishes cells from collagen fibers. In particular, this stain uses three different dyes: hematoxylin and acid fuchsine that colors in red both the cytoplasm and the nuclei, and blue aniline that colors in blue the collagen fibers (Figure 3). Induced hASCs, expanded by HFBR and stained with Alcian Blue, Masson’s trichrome and Safranin-O resulted positive for the presence of amorphous matrix components (Figures 3D, 3F), and for collagen fibers (Figure 3E). Same results have been obtained for hASCs expanded in flask (Figures 3L-3N). Undifferentiated samples resulted negative (Figures 3A-3C, 3G-3I). Histochemical analysis confirmed that chondrogenic differentiation has occurred, and apparently, the two methods of expansion do not affect the subsequent differentiation.
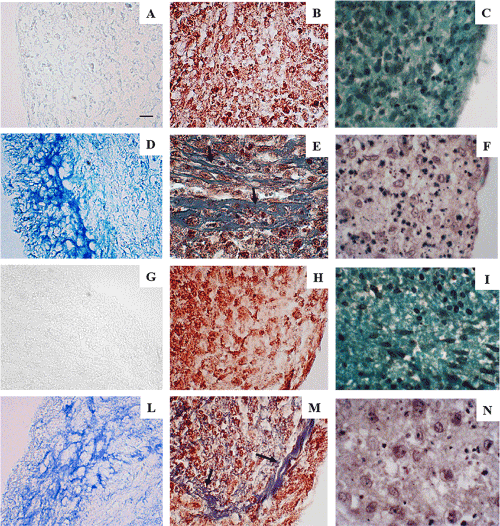
Figure 3. Representative optical microscopy photographs of differentiated and not differentiated hASCs. (A): Undifferentiated hASC spheroids expanded by HFBR stained with Alcian Blue, (B): Masson’s trichrome or (C): Safranin-O. (D): Induced hASC spheroids expanded by HFBR stained with Alcian Blue, (E): Masson’s trichrome or (F): Safranin-O. (G): Undifferentiated hASC spheroids expanded in flask are stained with Alcian Blue, (H): Masson’s trichrome or (I): Safranin-O. (L): Induced hASC spheroid expanded in flask stained with Alcian Blue, (M): Masson’s trichrome or (N): Safranin-O. Arrows indicated the presence of collagen fibrils in the ECM of the induced samples.
Transmission electron microscopy analysis
Pictures reported in Figure 4 are representative images of the ultrastructural features of control hASCs expanded by the two methods then maintained for 21 days in basal medium. The cell body is rounded, oval or polygonal and the plasma membrane is characterized by filopodia (f) and protrusions (not shown in these pictures). The nucleus (N) is large, indented, and often eccentric, with abundant euchromatin (*) and prominent nucleolus (n). Numerous ribosomes and polyribosomes together with lipid droplets (ld) are ubiquitously distributed in the cytoplasm. Extended endoplasmic reticulum (er) is composed by long, thin and irregular cisternae. Vesicles (v) and lysosomes (ly) of different size, containing low electron-dense material, are also present in the cytoplasm. Altogether, these characteristics suggest that hASCs have differentiated in preadipoblast cells. No differences were found between the two expansion methods.
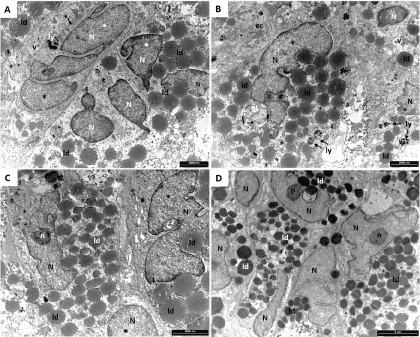
Figure 4. Representative TEM microphotographs of not induced hASCs expanded in HFBR (A,B) or in flask (C,D). The cells here presented shown the typical characteristic of preadipoblast. Indented nucleus (N) with euchromatin (*) abundant lipid droplets (ld), lysosomes (ly) and vesicles (v) are present. Extended endoplasmic reticulum (er) has been observed (B).
Figure 5 are representative images of the ultrastructural features of hASCs expanded by the two methods then cultured for 21 days in differentiating medium. The presence of abundant extracellular space (es) and the initial deposition of ECM compounds suggest that the differentiation process is taking place. The presence in the cells of a large N rich in euchromatin, the prominent n, the extended er, the Golgi apparatus (not shown in these figures), and extensive glycogen depots (gly), are indicative of substantial acceleration of the synthesis of proteins directed to extracellular space. Furthermore, the presence of several v containing homogeneous amorphous substance (ham), in close relationship with dense fibrillary material deposited upon the plasma membrane, suggest that the hASCs have differentiated in prechondroblast cells. In this phase of the differentiation, collagen fibrils (fi) appear distributed randomly throughout the extracellular space (Figures 5C-5D).

Figure 5. Representative TEM microphotographs of induced hASCs expanded in HFBR (A-C) or in flask (D). (A): A large nucleus (N) rich in euchromatin (*) and prominent nucleoli (n) are clearly visible. The extended endoplasmic reticulum (er) and the homogeneous amorphous substance (ham) are features typical of prechondroblast cells; vesicles (v) are also present. (B): glycogen depot (gly) and the ham together with the presence of abundant extracellular space (es) and initial deposition of extracellular matrix (ma) suggest that the differentiation process is taking place. Rare lipid droplets (ld) are also present. (C,D): Differentiation process is confirmed also by the presence of several fibrils (fi) in the es. Filopodia (f) are also present.
In Figures 6-7, the wide es is mainly occupied by well-organized collagen fi (Figures 6A-6B) and in Figures 6D and 7 is clear the presence of the characteristic banding. These matrix fi originated from dense aggregation of monomeric elements preformed inside the cortical ectoplasm then dispersed in groups into the ham and stored inside the v. Here, they grew up, maturated, folded and finally were released from the cell (Figures 7C). These last morphological characteristics are typical of chondroblast cells and confirm that the chondrogenic differentiation of hASCs has occurred. Also in this case, no differences were found between the two expansion methods. The different orientation of collagen fibrils is common in the early phase of cartilage development.
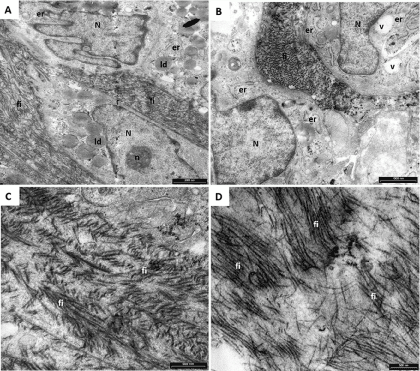
Figure 6. Representative TEM microphotographs of induced hASCs expanded in HFBR (A,C) or in flask (B,D). (A,B): The wide extracellular space is occupied by well-organized collagen fibrils (fi) indicating that differentiation to chondroblasts has occurred. Nucleus (N), nucleolus (n), endoplasmic reticulum (er), lipid droplets (ld) and vesicles (v) are indicated. (C,D): well- organized collagen fibrils indicate chondroblast differentiation.
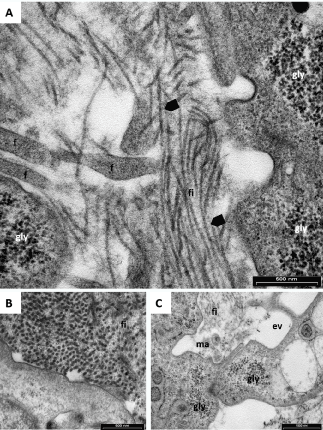
Figure 7. Representative TEM microphotographs of induced hASCs expanded in HFBR (A) or in flasks (B-C). (A): Presence of collagen banded fibrils (arrow heads), cell filopodia (f) and glycogen (gly) indicate that differentiation has occurred. (B): Transversal section of well-organized collagen fibrils (fi). (C): exocytic vesicle (ev) just opened, glycogen (gly), fibrils (fi) and extracellular matrix (ma).
Autologous chondrocyte implantation has been used as a strategy for the regeneration of damaged articular cartilage [40]. However, this cell-therapy approach is limited by critical drawbacks, such as donor site morbidity and loss of the chondrocyte phenotype [41]. Therefore, in recent years hASCs have emerged as valuable tool for the regeneration of articular cartilage based on their ability to undergo differentiation into the chondrogenic lineage [42]. The impact of orthopedic treatments based on hASCs is rapidly increasing and to date 20% of all clinical trials using hASCs are related to orthopedics (http://www.clinicaltrials.gov) as one of the main goals of stem cell therapy due also to the ease of access for treatment.
In our study, we aimed at comparing the chondrogenic differentiation ability of hASCs expanded in a HFBR or flask by using various complementary approaches: gene expression analysis, optical microscopy and TEM. Immunophenotyping showed that hASCs expanded in flask or HFBR retained a characteristic MSC immunophenotypic profile. In this sense hASCs from both cell expansion systems were remarkably similar, confirming data from the literature obtained with bone marrow-derived MSCs expanded in the same HFBR used in this study [43,44]. Concomitantly, the contamination of endothelial and hematopoietic cells was retained low enough to consider the cell therapy product for both cell expansion systems as pure [18]. Hence, both culture methods provided hASCs with of equal quality and purity that could be subjected to chondrogenic differentiation.
Gene expression analysis after differentiation showed how hASCs expanded in the HFBR had an upregulated average expression of SOX9, while the average expression in hASCs expanded in flask could not be considered as upregulated at this time-point. SOX9 acts as a crucial transcription factor involved in the chondrogenesis process and promotes the transcription of genes encoding the ECM molecules COL1A1, COL2A1, COL3A1 [45,46], ACAN [47], and COMP [48]. As observed for SOX9, also COMP, COL1A1 COL2A1, COL3A1, and ACAN expression for differentiated hASCs that originated from cell expansions in flask was lower when compared to HFBR-expanded cells. These genes follow specific expression kinetics during chondrogenesis, and the analysis of their mRNA levels at a single time-point is only a snapshot of the entire expression over time. Research groups who analyzed the gene expression of biomarkers during embryogenesis and chondrogenic induction of MSCs found that SOX9 expression starts at the mesenchymal osteochondroprogenitor stage and remains elevated in all differentiated chondrocytes until the cells reach hypertrophy [49]. Hamid and colleagues subjected hASCs to chondrogenic differentiation and observed a peak of mRNA level of COL2A1, ACAN, and COMP after 1 week of induction, while the peak of SOX9 was found at three weeks [50]. Xu and colleagues studied the temporal changes in chondrogenic genes and developed a staging scheme for in vitro differentiation of MSCs in three-dimensional alginate gels. The results of this study showed that SOX9 can be considered as an early marker of chondrogenesis whose expression peak is found at 6-12 days after induction, whereas COL2A1, ACAN, and COMP expression increased constantly during the induction [51]. On the other hand, Lin et al., reported about a sustained expression of SOX9, COL2A1, ACAN, and COMP mRNA up to 6 weeks of induction [52]. Taking all these studies together, there seems not to be an absolute consensus about the kinetics of expression of these marker genes, most probably due to the fact that research groups are using different experimental models of chondrogenesis, including different growth factors and cell types, thus making a direct comparison quite difficult.
For what concern our experiments, the results based only on gene expression analysis suggested that hASCs expanded in flask had a lower chondrogenic potential compared to HFBR expanded hASC. Since that, the molecular biology results may be not sufficiently informative, we have enforced the study with morphological analysis. Despite a certain variability, the results obtained by optical microscopy analysis after Alcian Blue, Safranin-O and Masson’s trichrome staining support the results of gene expression; hovewer, the presence of ECM components in all cell spheroids after 21 days of chondrogenic induction suggest that the differentiation process took place both for HFBR and flask expanded cells although with different efficiency and/or rate. TEM analysis validated the results obtained by optical microscopy and provided accurate details on cell differentiation and their capability to synthetize collagen fi in the es. In fact, the estrangement of the cells with the consequent appearance in the spheroid of wide es is indicative that differentiation is occurring. Furthermore, prominent n, the extended er and Golgi apparatus together with extensive gly depots, are representative of substantial acceleration of the synthesis of proteins directed to the es during chondrogenic differentiation [53-56]. The presence of a wide es invaded by well-organized collagen fi, even though differently oriented, is a further confirmation that prechondroblasts differentiated to chondroblasts. Also in this regard, no differences were apparent between the two expansion methods. As reported in the literature, during the early phase of cartilage development, there is a general arrangement of collagen fibrils that is concomitant with the increase in the proteoglycans content; consequently, the collagen fibers are differently oriented [57,58]. Overall, the application of a multidisciplinary approach has demonstrated to be effective for assessing cell differentiation. Since gene expression alone is clearly not informative enough to define the protein profile, considering that proteins are the ultimate effectors of biological function and are at the basis of cell phenotypes, we think that analysis of protein expression and cell morphology are more informative tools for assessing the extent of differentiation [59,60].
TEM demonstrated to be an excellent method to sustain qPCR for monitoring chondrogenic differentiation giving the opportunity to deeply investigate the presence of collagen fibers, as well as cell ultrastructure of chondrocytes. All together our results confirm that ultrastructural analysis by TEM, due to its high resolution, is a powerful morphological method able to give additional critical information.
The authors thank Lina Maciariello for technical assistance with cell culture, and Elise Roy, Stefano Balia, and Brent Rice of Terumo BCT for training and technical support with the Quantum Cell Expansion System. The authors also gratefully acknowledge “Centro Grandi Attrezzature per la Ricerca Biomedica” University of Insubria for instruments availability.
2021 Copyright OAT. All rights reserv
This study was supported by CARIPLO Foundation [grant number 2013-1052 to R.G.] and by FAR (Fondo comune di Ateneo per la Ricerca 2015) to Rosalba Gornati and Giovanni Berbardini.
The authors declare that they have no competing interest. The Swiss Stem Cell Foundation is owner of the patent “High safety process for the preparation of purified fraction” (PCT/2012/069261); however, this does not alter our adherence to Journal of Stem Cell Research and Medicine policy on sharing data and materials.
Cristina Pirrone: acquisition of data, analysis and interpretation of data, drafting the article
Alessandra Gobbetti: acquisition and analysis of data
Christian Caprara: analysis and interpretation of data, drafting the article
Giovanni Bernardini: conception and design, final approval of the version to be published
Rosalba Gornati: conception and design, drafting the article, final approval of the version to be published
Gianni Soldati: conception and design, final approval of the version to be published
- Vedicherla S, Buckley CT (2017) Cell-based therapies for intervertebral disc and cartilage regeneration- Current concepts, parallels, and perspectives. J Orthop Res 35: 8-22. [Crossref]
- Caplan AI (1991) Mesenchymal stem cells. J Orthop Res 9: 641-650. [Crossref]
- Bruder SP, Jaiswal N, Haynesworth SE (1997) Growth kinetics, self-renewal, and the osteogenic potential of purified human mesenchymal stem cells during extensive subcultivation and following cryopreservation. J Cell Biochem 64: 278-294. [Crossref]
- Friedenstein AJ, Gorskaja JF, Kulagina NN (1976) Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol 4: 267-274. [Crossref]
- Ashton BA, Abdullah F, Cave J, Williamson M, Sykes BC, et al. (1985) Characterization of cells with high alkaline phosphatase activity derived from human bone and marrow: preliminary assessment of their osteogenicity. Bone 6: 313-319. [Crossref]
- Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, et al. (2000) Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem 275: 9645-9652. [Crossref]
- Sekiya I, Larson BL, Vuoristo JT, Cui JG, Prockop DJ (2004) Adipogenic differentiation of human adult stem cells from bone marrow stroma (MSCs). J Bone Miner Res 19: 256-264. [Crossref]
- Dell'Accio F, De Bari C, Luyten FP (2001) Molecular markers predictive of the capacity of expanded human articular chondrocytes to form stable cartilage in vivo. Arthritis Rheum 44: 1608-1619. [Crossref]
- Kapur SK, Katz AJ (2013) Review of the adipose derived stem cell secretome. Biochimie 95: 2222-2228. [Crossref]
- Casado JG (2014) Immunomodulatory potential of human adipose mesenchymal stem cells derived exosomes on in vitro stimulated T cells. Front Immunol 5: 1-9. [Crossref]
- Mattar P, Bieback K (2015) Comparing the Immunomodulatory Properties of Bone Marrow, Adipose Tissue, and Birth-Associated Tissue Mesenchymal Stromal Cells. Front Immunol 6: 560. [Crossref]
- Krampera M, Galipeau J, Shi Y (2013) Immunological characterization of multipotent mesenchymal stromal cells-The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy 15: 1054-1061. [Crossref]
- Hass R, Kasper C, Böhm S, Roland J (2011) Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Communication and Signaling. BioMed Central 9: 12. [Crossref]
- Cherubino M, Valdatta L, Balzaretti R, Pellegatta I, Rossi F, et al. (2016) Human adipose-derived stem cells promote vascularization of collagen-based scaffolds transplanted into nude mice. Regen Med 11: 261-271. [Crossref]
- Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, et al. (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13: 4279-4295. [Crossref]
- De Ugarte DA, Morizono K, Elbarbary A, Alfonso Z, Zuk PA, et al. (2003) Comparison of multi-lineage cells from human adipose tissue and bone marrow. Cells Tissues Organs 174: 101-109. [Crossref]
- Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J (2012) Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev 21: 2724-2752. [Crossref]
- Bourin P, Bunnell BA, Casteilla L, Dominici M, Katz AJ, et al. (2013) Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: a joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy 15: 641-648. [Crossref]
- Lambrechts T, Papantoniou I, Rice B, Schrooten J, Luyten FP, et al. (2016) Large-scale progenitor cell expansion for multiple donors in a monitored hollow fibre bioreactor. Cytotherapy 18: 1219-1233.
- Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU (1998) In Vitro Chondrogenesis of Bone Marrow-Derived Mesenchymal Progenitor Cells. Exp Cell Res 238: 265-272. [Crossref]
- Woods A, Wang G, Beier F (2007) Regulation of chondrocyte differentiation by the actin cytoskeleton and adhesive interactions. J Cell Physiol 213: 1-8. [Crossref]
- Barry F, Boynton RE, Liu B, Murphy JM (2001) Chondrogenic differentiation of mesenchymal stem cells from bone marrow: differentiation-dependent gene expression of matrix components. Exp Cell Res 268: 189-200. [Crossref]
- Estes BT, Wu AW, Guilak F (2006) Potent induction of chondrocytic differentiation of human adipose-derived adult stem cells by bone morphogenetic protein 6. Arthritis Rheum 54: 1222-1232. [Crossref]
- Biddulph DM, Sawyer LM, Smales WP (1984) Chondrogenesis of chick limb mesenchyme in vitro. Effects of prostaglandins on cyclic AMP. Exp Cell Res 153: 270-274. [Crossref]
- VanGuilder HD1, Vrana KE, Freeman WM (2008) Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 44: 619-626. [Crossref]
- Vogel C, Marcotte EM (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13: 227-232. [Crossref]
- Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, et al. (2004) Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics 3: 960-969. [Crossref]
- Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, et al. (2010) Defining the transcriptome and proteome in three functionally different human cell lines. Mol Syst Biol 6: 450. [Crossref]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, et al. (2011) Global quantification of mammalian gene expression control. Nature 473: 337-342. [Crossref]
- Vogel C, Abreu Rde S, Ko D, Le SY, Shapiro BA, et al. (2010) Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol 6: 400. [Crossref]
- Kendrick N (2014) A gene's mRNA level does not usually predict its protein level. Kendrick Laboratories.
- Chatterjee S, Roy A, Laskar A (2012) Electron microscopy in the perspective of modern biology: Ultravision and ultradimension. Current Microscopy Contributions to Advances in Science and Technology 2: 891-902.
- Minonzio G, Corazza M, Mariotta L, Gola M, Zanzi M, et al. (2014) Frozen adipose- derived mesenchymal stem cells maintain high capability to grow and differentiate. Cryobiology 69: 211-216. [Crossref]
- Ragni E, Viganò M, Rebulla P, Giordano R, Lazzari L (2013) What is beyond a qRT-PCR study on mesenchymal stem cell differentiation properties: how to choose the most reliable housekeeping genes. J Cell Mol Med 17: 168-180. [Crossref]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, et al. (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8: 315-317. [Crossref]
- Hardingham TE, Oldershaw RA, Tew SR (2006) Cartilage, SOX9 and Notch signals in chondrogenesis. J Anat 209: 469-480. [Crossref]
- Dudhia J (2005) Aggrecan, aging and assembly in articular cartilage. Cell Mol Life Sci 62: 2241-2256. [Crossref]
- Newton G, Weremowicz S, Morton CC, Copeland NG, Gilbert DJ, et al. (1994) Characterization of human and mouse cartilage oligomeric matrix protein. Genomics 24: 435-439. [Crossref]
- Burgeson RE, Nimni ME (1992) Collagen types. Molecular structure and tissue distribution. Clin Orthop Relat Res 250-272. [Crossref]
- Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, et al. (1994) Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med 331: 889-895. [Crossref]
- Giannini S, Buda R, Cavallo M, Ruffilli A, Cenacchi A, et al. (2010) Cartilage repair evolution in post-traumatic osteochondral lesions of the talus: from open field autologous chondrocyte to bone-marrow-derived cells transplantation. Injury 41: 1196-1203. [Crossref]
- Im GI (2016) Regeneration of articular cartilage using adipose stem cells. J Biomed Mater Res A 104: 1830-1844. [Crossref]
- Rojewski MT, Fekete N, Baila S, Ruffilli A, Cenacchi A, et al. (2013) GMP-Compliant Isolation and Expansion of Bone Marrow-Derived MSCs in the Closed, Automated Device Quantum Cell Expansion System. Cell transplant 22: 1981-2000. [Crossref]
- Lechanteur C (2014) Large-Scale Clinical Expansion of Mesenchymal Stem Cells in the GMP-Compliant, Closed Automated Quantum® Cell Expansion System: Comparison with Expansion in Traditional T-Flasks. J Stem Cell Res Ther 04: 1-11.
- Bell DM, Leung KK, Wheatley SC, Ng LJ, Zhou S, et al. (1997) SOX9 directly regulates the type-II collagen gene. Nat Genet 16: 174-178. [Crossref]
- Lefebvre V, Huang W, Harley VR, Goodfellow PN, De Crombrugghe B. (1997) SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol 17: 2336-2346. [Crossref]
- Sekiya I, Tsuji K, Koopman P, Watanabe H, Yamada Y, et al. (2000) SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J Biol Chem 275: 10738-10744. [Crossref]
- Liu C-J, Zhang Y, Xu K, Parsons D, Alfonso D, et al. (2007) Transcriptional activation of cartilage oligomeric matrix protein by Sox9, Sox5, and Sox6 transcription factors and CBP/p300 coactivators. Front Biosci 12: 3899-3910. [Crossref]
- Zhao Q, Eberspaecher H, Lefebvre V, De Crombrugghe B (1997) Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev Dyn 209: 377-386. [Crossref]
- Hamid AA, Idrus RB, Saim AB, Sathappan S, Chua KH (2012) Characterization of human adipose-derived stem cells and expression of chondrogenic genes during induction of cartilage differentiation. Clinics (Sao Paulo) 67: 99-106. [Crossref]
- Xu J, Wang W, Ludeman M, Cheng K, Hayami T, et al. (2008) Chondrogenic differentiation of human mesenchymal stem cells in three-dimensional alginate gels. Tissue Eng(PartA) 14: 667-680. [Crossref]
- Lin Y, Luo E, Chen X, Liu L, Qiao J, et al. (2008) Molecular and cellular characterization during chondrogenic differentiation of adipose tissue-derived stromal cells in vitro and cartilage formation in vivo. J Cell Mol Med 9: 929-939. [Crossref]
- Godman GC, Porter KR (1960) Chondrogenesis, studied with the electron microscope. J Biophys Biochem Cytol 8: 719-760. [Crossref]
- Daimon T (1977) The presence and distribution of glycogen particles in chondrogenic cells of the tibiotarsal anlage of developing chick embryos. Calcif Tissue Res 23: 45-51. [Crossref]
- Yabu M, Takaoka K, Hashimoto J, Fujita H (1992) Immunohistochemical, autoradiographic and electron microscopic studies on the transformation of fibroblasts into chondrocytes in the mouse subfascia induced by bone morphogenetic protein. Histochemistry 97: 463-468.
- Manea CM, Rusu MC, Constantin D, Manoiu VM, Moldovan L, et al. (2014) Ultrastructural features of human adipose-derived multipotent mesenchymal stromal cells. Rom J Morphol Embryol 55: 1363-1369. [Crossref]
- ap Gwynn I, Wade S, Ito K, Richards RG (2002) Novel aspects to the structure of rabbit articular cartilage. Eur Cell Mater 4: 18-29. [Crossref]
- Julkunen P, Iivarinen J, Brama PA, Arokoski J, Jurvelin JS, et al. (2010) Maturation of collagen fibril network structure in tibial and femoral cartilage of rabbits. Osteoarthritis and Cartilage 18: 406-415. [Crossref]
- Li Z, Hu S, Ghosh Z, Han Z, Wu JC, et al. (2011) Functional Characterization and Expression Profiling of Human Induced Pluripotent Stem Cell- and Embryonic Stem Cell-Derived Endothelial Cells. Stem Cells Dev 20: 1701-1710. [Crossref]
- Burattini S, Falcieri E (2013) Analysis of cell death by electron microscopy. Kimberly McCall and Charles Klein (eds). Springer Science+Business Media LLC New York, USA, pp: 77-89.