A normal human karyotype contains 22 pairs of autosomal chromosomes and one pair of sex chromosomes which are denoted 46, XX for women, and 46, XY for men. An aneuploid karyotype contains an abnormal number of chromosomes that may apply to any of the 22 autosomal pairs, or the sex chromosomes. Overall, excess in autosomal chromosomes is lethal, with the exception of chromosomes 13, 18 and 21. Individuals with such aneuploidies have complex, severe syndromes. Accordingly, having abnormal number of chromosomes in the embryo is determined by the first and second trimester pregnancy surveys [1]. In contrast, although identifiable through Noninvasive Prenatal Testing (NIPT) and amniocentesis, sex chromosome aneuploidy is harder to detect with these surveys [2]. Remarkably, the risk for autosomal aneuploids increases with women age, however for abnormal number of ChrX it is age independent and show a constant risk [3]. Generally speaking, the aneuploidy of X chromosome results in relatively mild phenotypes (Table 1). This is supported by the fact that the only viable human karyotype with a complete chromosomal loss occurs in females with Turner Syndrome (TS; 45, X; Table 1). In this report, we discuss the molecular basis underlying the phenotype variability and the apparent tolerance associated with ChrX.
Table 1: Sex chromosome aneuploids in human population.
45, X - Turner Syndrome (TS) females have only one ChrX. Symptoms include short stature, development delays, delayed puberty, infertility, learning disabilities or behavior problems, premature ovarian failure, a webbed neck and lymphedema of the limbs. Some women have skeletal abnormalities, kidney problems, and/or a congenital heart defect. The occurrence is 1:2500 newborn females [19]. |
47, XXX - Triple X chromosome females have ChrX trisomy, and are mostly asymptomatic. Symptoms include tall stature, some learning disabilities and various psychopathology. The occurrence is 1:1000 newborn females [20]. |
47, XXY - Klinefelter syndrome (KS) males having two copies of ChrX and a Y chromosome. Most men are infertile due to hypogonadism and/or cryptorchidism. Some might have gynecomastia, tall stature and some learning disabilities. The occurrence is 1:660 newborn boys [21]. |
Rare aneuploidy of females and males with more than 3 copies of ChrX are also viable (e.g., 48, XXYY, 48, XXXX 48, XXXY, 49, XXXXY etc). These karyotypes are very rare and often appear as chromosomal mosaicism. In general, the more severe symptoms are associated with a larger number of copies. |
Unique factors characterize the inheritance and genetics of the sex chromosomes (ChrX and ChrY). Significantly, the two sex chromosomes differ from each other in almost all quantitative properties. Specifically, ChrX is 2.7 times longer than ChrY, and has 13 times more coding genes relative to ChrY. Does it mean that women (46, XX) have many more expressed genes than men (46, XY)? Well, not quite. During female embryogenesis, already at the blastocyst stage, a lifetime decision for inactivating one of two X chromosomes is made in each cell of the developing embryo (Figure 1a). The decision, once made, dictates the identity of the inactivated ChrX being maternal or paternal for the entire cell lineage of that cell. As a result, from the point of view of gene expression, each cell in the women body has one active X (Xa) and one inactive X (Xi) chromosome. The master regulator of this cell decision is a noncoding gene called XIST, which together with other noncoding RNAs, drives the ChrX-inactivation process. This non-reversible cell decision involves a tight packing of one of the X-chromosomes (Xi) which becomes predominantly silent.
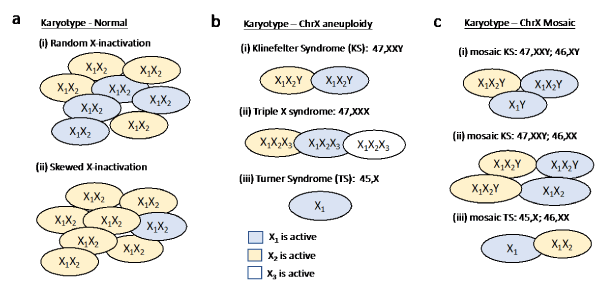
Figure 1. Karyotypic diversity of ChrX according to the pattern of X-inactivation.
Importantly, X-inactivation process involves high precision step of ChrX “counting”. Namely, in any cell with more than one ChrX copy (e.g., normal females, Triple X chromosome or Klinefelter syndrome, Figure 1b), only one of the ChrX remains active, while the rest are inactivated. Keeping this in mind, a phenotype diversity that is associated with polyploidy of ChrX is unexpected. In reality, many clinical studies show that alteration in ChrX ploidy results in a wide range of phenotypes (Table 1). In this report, we introduce numerous explanations for the observed phenotype variability associated with ChrX. We focus on the molecular basis of such variability as studied in humans with normal and altered karyotypes.
Overall, the phenotype and clinical variability (e.g. stature) of any individual is driven by an integration of the expressed genes, and the identity of the expressed copy in all the cells of that individual. The case of ChrX is unique because of the random decision that determines which of the two ChrX remains active (X1 or X2, Figure 1a). At the organism level, the number of cells carrying X1 or X2 is expected to be equal. Still locally, cells carrying the same ChrX copy appear in patches. Figure 1a illustrates scenarios of X-inactivation through the local cellular compositions. The phenomenon in which a symmetry between cells expressing genes from X1 or X2 is missing (Figure 1a), and most cells express genes from only one of the X chromosomes is called ChrX skewing. For example, consider a ChrX gene that is critical for brain development with a mutation in one of the ChrX copies. In this case, the choice of which ChrX is inactivated is critical for brain development in females. Indeed, in many cases, only cells that express the normal copy will survive, leading to a skewing appearance (Figure 1a, ii). A biased selection of a specific copy (benign or pathogenic) is the basis for a wide range of clinical outcomes.
Additionally, a few dozen of genes do not adhere to the X-inactivation process. These genes are called escapees and are expressed from both ChrX copies, albeit not always to the same levels. In most instances, the expression from Xi is suppressed with respect to Xa. The status of gene as escapees is crucial for phenotypes that depend on the amounts of specific gene products [4]. Indeed, monitoring the expression levels of thousands of cells with normal karyotypes indicated variability across cells, tissues, organs and individuals [5].
In order to assess the factors affecting ChrX phenotype diversity, ChrX genes are grouped according to their X-inactivation status [6]. For each genes’ group, we exemplify a representative gene for illustrating the connection between the phenotype and the gene status in normal and altered karyotypes.
The first group includes genes from regions at the tips of ChrX that are shared between ChrX and ChrY and are called Pseudoautosomal Regions (PAR). As their name implies, The PAR genes are not inactivated and are expressed from both copies as autosomal genes do. Generally, when considering normal karyotypes, PAR genes contribute equally to the phenotype in males and females, irrespective of their origin from ChrX or ChrY. Altogether there are 28 PAR genes. The SHOX (short stature homeobox-containing) is a PAR gene that is involved in the complex phenotype of body growth. It was found as a growth developmental causal gene by a genomic analysis of 36 individuals with short stature and their normal family relatives [7]. Following the PAR gene expression pattern, the stature phenotype is expected to be correlated with the number of ChrX and ChrY copies. Indeed, the restricted expression of SHOX in Turner Syndrome females (TS, 45, X) leads to a short stature, while its overexpression in Triple-X syndrome (47, XXX) and Klinefelter Syndrome (KS, 47, XXY) leads to their tall stature (Table 1).
A second group concerns the inactivated genes that account for the majority of the ChrX genes. As the inactivated genes are expressed from a single copy in both males and females, they are not expected to contribute much to the phenotype variability of polyploids. An example of an inactivated gene is the enzyme G6PD (Glucose-6-phosphate dehydrogenase). Mutations in this gene cause the most common human enzyme defect [8]. Due to the stochastic X-inactivation process, half of the cells in females still produce functional G6PD which suffices the needs for the functional enzyme. Still, some females are severely affected despite having only one mutated G6PD copy [9]. Such phenotypic variability is a result skewing (Figure 1a). Phenotype variability can also be a result of a partial relief from ChrX inactivation, a phenomenon that was associated with aged population [10].
The less defined group includes a handful of genes that escape X-inactivation and thus express from both ChrX copies. As oppose to the PAR genes, they are not expressed from ChrY [11]. Many of these genes are involved in fundamental processes of the cell [12]. This group can be divided into two sets: the smaller fraction exhibits an escaping characteristic across all conditions, cell types and individuals (refer to consistent escapees), while the major fraction is signified by genes’ ability to escape X-inactivation only in some settings but not in others [6, 13]. However, the principles by which genes become inactivated or escapee in different settings are poorly understood. We illustrate the clinical phenotype associated with consistent and variable escapees. An example of a consistent escapee gene is KDM6A. This gene is a tumor suppressor, with recurrent mutations in prostate cancer, medulloblastoma and renal carcinoma. Women bearing mutations in one copy of the gene exhibit a phenotype whose severity reflects the expression of the mutated KDM6A from the Xi copy. In general, the expression of escapees in females underlies some of the gender dependent bias in variety of tumor types [14].
Of a special importance for the phenotype variability is the group of variable escapee genes. For example, OFD1 (oral facial-digital type 1) is an essential gene, and its absence leads to male lethality. The affected females express a broad clinical variability that includes different degree of mental retardation, polycystic kidney disease, malformations of the face, oral cavity and digits. Importantly, such variability in phenotypes appears even within family members that share the same mutation. In general, variable escapees lead to a clinical variability not only among individuals, but also across cells, cell types, tissues and organs [15]. Moreover, a nonrandom X-inactivation of OFD1 add to the phenotype variability. Specifically, in 30% of the patients, a preferred expression from the normal allele was observed (Figure 1b) [16]. Many escapee genes are associated with mental retardation, disruptions in cognitive function and emotional development [17]. Therefore, it may underlie the abundance of such phenotypes in various poly-X karyotypes (e.g., Triple X-syndrome, Figure 1b, ii).
Combining the paradigm of “ChrX counting” with the X-inactivation genes’ groups allows revisiting the molecular source of phenotype variability in altered karyotypes (Table 1). The most common sex chromosome alterations are associated with KS (47, XXY), TS (45, X) and Triple X syndrome (47, XXX). In these cases, an additional source of genetic variation needs to be considered. During cell divisions stochastic mistakes can result in a mosaic cell population (Figure 1c). Namely, some cells exhibit an aneuploid karyotype while other cells carry normal karyotypes. It is plausible that sex chromosome mosaicism may attenuate the severity of the clinical outcomes. Actually, most TS cases are mosaic, with the most dominant variant composed of 45, X and normal (46, XX) cells (Figure 1c, iii). As anticipated, in KS and TS individuals, higher proportion of cells with normal karyotypes correlates with less severe phenotypes (Table 1). As the aneuploidy in ChrX results in elevated expression of escapees and PAR genes, an impact on the clinical manifestation is anticipated. In accordance with this assumption, KS is associated with a 20-fold higher risk of developing breast cancer when compared to normal males. The link to many comorbidities was also attributed to the overexpression of escapees and PAR genes [18].
To sum up, genetic processes of X-inactivation and escaping from it, choices of specific ChrX copy, the degree of bias in the cellular ChrX identity and the cellular composition of a mosaic karyotype, together contribute to the clinical and phenotypic outcomes. We presented numerous ways affecting gene expression levels that are unique to ChrX. It enables the affected individuals to survive with only mild clinical outcomes by tolerating the adverse outcome of pathogenic mutations that occur in only one of the ChrX copies. Understanding the properties of ChrX genes as a source for phenotypes and clinical variability has the potential to impact medical practice and prenatal testing in cases of sex chromosome aneuploidy (Table 1).
References
- Wilton L (2002) Preimplantation genetic diagnosis for aneuploidy screening in early human embryos: a review. Prenat Diagn 22: 512-518. [Crossref]
- Viuff MH, Stochholm K, Uldbjerg N, Nielsen BB, Danish Fetal Medicine Study G, et al. (2015) Only a minority of sex chromosome abnormalities are detected by a national prenatal screening program for Down syndrome. Hum Reprod 30: 2419-2426.
- Nicolaides KH (2011) Screening for fetal aneuploidies at 11 to 13 weeks. Prenat Diagn 31: 7-15. [Crossref]
- Deng X, Berletch JB, Nguyen DK, Disteche CM (2014) X chromosome regulation: diverse patterns in development, tissues and disease. Nat Rev Genet 15: 367-378. [Crossref]
- Tukiainen T, Villani AC, Yen A, et al. (2017) Landscape of X chromosome inactivation across human tissues. Nature 550: 244-248. [Crossref]
- Balaton BP, Cotton AM, Brown CJ (2015) Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol Sex Differ 6: 35. [Crossref]
- Rao E, Weiss B, Fukami M, Rump A, Niesler B, et al. (1997) Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 16: 54-63. [Crossref]
- Cappellini MD, Fiorelli G (2008) Glucose-6-phosphate dehydrogenase deficiency. Lancet 371: 64-74. [Crossref]
- Au WY, Lam V, Pang A, Lee WM, Chan JL, et al. (2006) Glucose-6-phosphate dehydrogenase deficiency in female octogenarians, nanogenarians, and centenarians. J Gerontol A Biol Sci Med Sci 61: 1086-1089. [Crossref]
- Machiela MJ, Zhou W, Karlins E, Sampson JN, Freedman ND, et al. (2016) Female chromosome X mosaicism is age-related and preferentially affects the inactivated X chromosome. Nat Commun 7: 11843. [Crossref]
- Carrel L, Willard HF (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434: 400-404. [Crossref]
- Berletch JB, Yang F, Disteche CM (2010) Escape from X inactivation in mice and humans. Genome Biol 11: 213. [Crossref]
- Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, et al. (2018) Corrigendum: Landscape of X chromosome inactivation across human tissues. Nature 555: 274. [Crossref]
- Dunford A, Weinstock DM, Savova V, Schumacher SE, Cleary JP, et al. (2017) Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat Genet 49: 10-16. [Crossref]
- Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, et al. (2013) Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol 30: 2588-2601. [Crossref]
- Morleo M, Franco B (2008) Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet 45: 401-408. [Crossref]
- Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF (2002) Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am J Hum Genet 71: 168-173. [Crossref]
- Belling K, Russo F, Jensen AB, Dalgaard MD, Westergaard D, et al. (2017) Klinefelter syndrome comorbidities linked to increased X chromosome gene dosage and altered protein interactome activity. Hum Mol Genet 26: 1219-1229. [Crossref]
- Pinsker JE (2012) Clinical review: Turner syndrome: updating the paradigm of clinical care. J Clin Endocrinol Metab 97: E994-1003. [Crossref]
- Otter M, Schrander-Stumpel CT, Curfs LM (2010) Triple X syndrome: a review of the literature. Eur J Hum Genet 18: 265-271. [Crossref]
- [Crossref] Bojesen A1, Gravholt CH (2007) Klinefelter syndrome in clinical practice. Nat Clin Pract Urol 4: 192-204.