Abstract
Purpose: The purpose of this study was to screen for mutations within mitochondrial creatine kinase (CKMT) genes, which encode for isoenzymes critical for high energy metabolism, such as that found in retina.
Methods: DNA was extracted of lymphocytes from clinically characterized age-related macular degeneration (AMD) patients (n=71). Flanking primers were used to polymerase chain reaction (PCR) amplify and sequence the exons, the open reading frame, and promoter regions of the CKMT1A, CKMT1B and CKMT2 genes. An additional 72 individuals with AMD were screened for the novel CKMT2 mutation in exon 4 (NM_001825.2 c.274C>T) by PCR and enzyme digestion. The RNA was extracted from cultured human retinal pigment epithelial cells (ARPE-19) and reverse transcript-PCR (RT-PCR) was performed with primers for the CKMT2 gene.
Results: With our screening procedure, we identified 5 DNA variants in the CKMT2 gene, 2 of which were novel (NM_001825.2 c.274C>T; 92Q>X, Exon 4 and NM_001825.2.-498G>GA; 5’UTR, Exon 1). The putative termination mutation in the CKMT2 gene (NM_001825.2 c.274C>T; 92Q>X) was in a proband individual but lacking in his unaffected son and 142 unrelated AMD patients. Cultured human RPE cells can express the sarcomeric mitochondrial CKMT2 gene product.
Conclusions: This is the first report of a termination mutation within the human CKMT2 gene, which is critical for transfer of high-energy phosphate from mitochondria to creatine, a cytosolic carrier. Clinically, the subject had AMD, exercise fatigue, atrial fibrillation and deafness, all of which are known to be related to mitochondrial dysfunctions. Based upon our finding, we propose this CKMT2 mutation may be a candidate gene for the phenotypes that include this quartet of symptoms.
Key words
CKMT2, fatigue, age-related macular degeneration, deafness, atrial fibrillation, CKMT1A, CKMT1B
Abbreviations
AMD: Age-Related Macular Degeneration; CKMT: Creatine Kinase; PCR: Polymerase Chain Reaction; ARPE-19: Human Retinal Pigment Epithelial Cell Line; RT-PCR: Reverse Transcript-PCR; mtDNA: Mitochondrial DNA; RPE: Retinal Pigment Epithelial; CK: Creatine Kinase; CT: Computer Tomography; CKMT2: Mitochondrial Creatine Kinase 2
Introduction
Organs such as brain, heart, muscle and retina have significantly higher energy demands as compared to other organs. Therefore, mutations or sequence variations in genes that contribute to energy generation through oxidative phosphorylation will have significant effects on the function of these organs.
Retinal tissues are extremely metabolically active and have numerous mitochondria in the inner segment of the photoreceptor cells. There is increasing evidence that alterations in the mitochondrial DNA (mtDNA) and in mitochondrial function may play a role in age-related macular degeneration (AMD), a retinal disorder that is a leading cause of vision loss in people over 60 years of age [1-8]. Atrophic AMD is a severe form of the disease that exhibits loss of the retinal pigment epithelial (RPE) cells and overlying photoreceptors. It has been reported that cultured RPE cells that are mitochondria deficient show an altered expression of creatine kinase (CK) genes [9]. These CK genes function to catalyze the transfer of high energy phosphates between ATP and creatine, thereby yielding ADP and phosphocreatine. CKMT1A and CKMT1B are ubiquitous mitochondrial genes that are on human chromosome 15q15.3, differ by only 2 nucleotides and code for an identical CKMT1 protein. The CKMT2 gene is located on human chromosome 5q13.3 and encodes for a sarcomeric mitochondrial creatine kinase isoenzyme. There are 3 transcript variants (NM_001825, NM_001099735 and NM_001099736) of the CKMT2 gene, all of which encode the same 419 aa protein and have similar functions but are expressed in different tissues. All the CKMT genes are critical for energy generation.
In the present study, we initially screened the CKMT1A, CKMT1B and CKMT2 genes from individuals with clinically significant AMD because these genes are highly conserved and critical for tissues with high energy requirements, such as the retina. Therefore, any variations in these genes could cause significant dysfunction and pathology for the effected retinas. With our screening procedure, we identified 5 DNA variants in the CKMT2 gene, 2 of which were novel (NC.000005.10; hg19:chr5:81,252,816.274C>T; 92Q>X, exon 4 and hg19:chr5:81,233,356.498G>GA, exon 1). One proband subject had the putative termination mutation in the CKMT2 gene (NM_001825.2) and thorough review of his medical status showed that in addition to AMD, he also had exercise fatigability, deafness and atrial fibrillation.
Materials and methods
The study was approved by the institutional review boards of University of California Irvine (HS# 2003-3131) and Cedars-Sinai Medical Center (IRB #1708). Informed consents were obtained from participants and the study performed according to the tenets of the Declaration of Helsinki for research involving human subjects.
AMD Classification
One hundred forty-three subjects underwent a complete dilated ophthalmic examination by Board certified ophthalmologists (D.S.B., A.B.N., and M.C.K.) including both slit lamp examination and an indirect ophthalmic exam with a 90 diopter lens or a fundus contact lens. Fundus photos, fluorescence and/or indocyanine green angiography were performed. The photos and angiograms were read by masked graders who were board certified retinal specialists. The subjects were graded according to the Clinical Age-Related Maculopathy Staging System (CARMS) [10]. Grade 3 had large soft drusen or several intermediate size drusen or drusenoid retinal pigment epithelial detachment and for this study is referred to as Early AMD. In this study the term Late AMD is the combination of Grade 4 which is geographic atrophy and grade 5 which is neovascular or serous exudative AMD. No stage 1 or 2 AMD patients were included in this study. Upon analyses, a putative termination heterozygous mutation NM_001825.2 c.274C>T (92Q>X) was found in one individual that we designated AMD-02-141 and further analyses of the proband subject and his son were conducted.
Clinical description of proband patient
Past ocular history: The proband subject is an 87 year old Caucasian male that has been treated for 6 years for the wet form of AMD. His ocular history is significant for cataract surgery for both eyes and YAG laser capsulotomy for his right eye in 1997. In January 2005, he developed a subretinal membrane in the left eye which was treated with photodynamic therapy and an intravitreal injection of KenalogÔ. In July 2006, his left eye received another treatment of photodynamic therapy combined with an AvastinÔ intravitreal injection. Subsequently, he has developed dry AMD in the right eye and had best corrected visual acuity of 20/400 in both eyes. Dilated exam shows large macular glial scars with atrophy and age-related pigment alterations of the retinas (Figure 1).
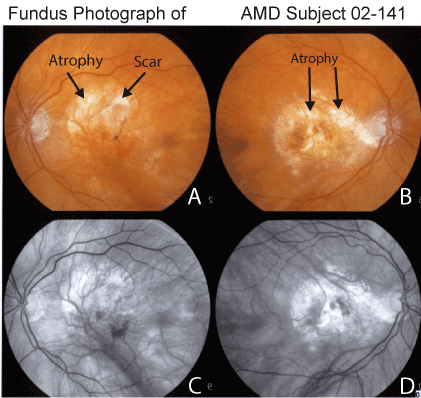
Figure 1. Fundus photography of the macular region of patient AMD-02-141 showing large area of macular atrophy, subretinal glial scars and pigmentary alterations of the retina (Panel A, left eye and Panel B, right eye). Red free photographs demonstrate regions with loss of retinal pigment epithelium throughout the macula of both eyes (Panel C, left eye and Panel D, right eye).
Past medical history: The proband has loss of hearing (presbycusius), hyperlipidemia, chronic sinusitis, and benign prostatic hypertrophy. He also has experienced loss of exercise tolerance and becomes easily fatigued which caused him to give up the use of the rowing machine and his stationary bicycle training. The proband has a common form of atrial flutter of indeterminate duration and an EKG has shown atrial flutter with rapid ventricular response that inverted to atrial fibrillation with controlled ventricular response. In 1982, he was diagnosed with a history of paraoxysmal atrial flutter. A stress echo exam showed no ischemia, occasional premature ventricular complexes and moderately frequent premature atrial complexes. In 1999, his chronic sinusitis was evaluated and serial axial computer tomography (CT) scan of the paranasal sinuses showed marked mucosal disease in the left and to a lesser extent right anterior and posterior ethmoids. He had mucosal thickening in the maxillary sinuses (left>right), the periphery of the middle and inferior meati and the nasal septum along with an anterior nasal septal defect.
Past surgical history: His surgical history includes previous sinus surgery, vasectomy, extraction of kidney stones, appendectomy, bilateral osteomeatal surgery and herniorrhaphy.
Database accession numbers
Human reference sequences used for primer design and sequence analysis were accessed at NCBI and the accession numbers are as follows. CKMT1a: NM_001015001.1; CKMT1b: NM_020990.3; CKMT2: NM_001825.2, NM_001099735.1, and NM_001099736.1.
Extraction of total DNA
Venous blood samples (10 ml) from AMD patients were collected in tubes with 10mM ethylenediaminetetra-acetic acid (n=143). The Gentra Puregene DNA (Qiagen, Inc., Valencia, CA) extraction kit was used to isolate total DNA from leukocytes.
Sequencing
The sequencing primers used for the CKMT1A, CKMT1B and CKMT2 are listed in Table 1. Primers were designed using PRIMER3 program (Whitehead Institute for Biomedical Research). All sequenced PCR products were treated to digest unused primers and nucleotides (ExoSAP-IT, USB Corp., Cleveland, OH). Sequencing was performed at the UCLA Sequencing and Genotyping Core facility. Sequence analyses were performed using the Mutation Surveyor program, (Softgenetics, State College, PA). The DNA from 71 subjects underwent sequencing of all 11 exons of the CKMT2 gene. In addition, the DNA from an additional 72 individuals with AMD were screened for the novel CKMT2 mutation in exon 4 (NM_001825.2 c.274C>T) by PCR and enzyme digestion with Xba1 restriction enzyme (NEB, Ipswich, MA). The products were run on a 1.5% agarose gel stained with ethidium bromide.
Cell culture
ARPE-19 cells (
ATCC
, Manassa, VA) were grown in 1:1 mixture (vol/vol) of Dulbecco’s modified Eagle’s and Ham’s nutrient mixture F-12; (Invitrogen-Gibco, Carlsbad, CA), nonessential amino acids 10mM 1×, 0.37% sodium bicarbonate, 0.058% L-glutamine, 10% fetal bovine serum, and antibiotics (penicillin G 100U/ml, streptomycin sulfate 0.1 mg/ml, gentamicin 10 µg/mL, amphotericin-B 2.5 µg/mL). Cells were plated into 60mm dishes and incubated at 37oC in 5% CO2 until confluent.
RNA isolation
Human ARPE-19 cell cultures were rinsed in phosphate buffered saline (PBS, pH 7.2) and RNA was isolated using the RNeasy Extraction kit (Qiagen) following the manufacturer’s protocol. Blood was obtained from patient AMD-02-141 and his son and the RNA was isolated using the Versagene Blood Kit (5 Prime, Gaithersburg, MD). One microliter of the RNA was analyzed for quantity using the NanoDrop (Thermo Scientific, Wilmington, DE).
cDNA synthesis and reverse-transcribed-PCR (RT-PCR)
All RNA samples were reverse transcribed into cDNA using the SMARTer kit (Clontech, Mountain View, CA). The cDNA from the ARPE-19 cells was amplified (Qiagen) with CKMT2 primers (Table 1), annealing temperature of 67°C and PCR products were run on a 1% agarose gel and stained with ethidium bromide.
Primer Sequence Forward Reverse |
CKMT2 |
|
|
CKMT2E1 |
cggaggtctacgctgggattggtgagg |
tcctccaaggcccttcacactggctct |
CKMT2E2 |
caggcataagccaccatgcccagt |
ctcatcccaagaacacagaggcccaca |
CKMT2E3 |
tggctcctgcagttctcttgcccattg |
tctcttgacctcgtgatctggccttcca |
CKMT2E4 |
gggagcctagtggaaggatgggcaaaca |
gccccatagagaagaaaaggaccccagga |
CKMT2E5 |
acaaggggcaagtgaatggaaaggtct |
gggcagtcacagtgctttccaggggta |
CKMT2E6 |
tggcgattagaacccactgtggctgtg |
aaggactgagcagagcaccaggagagg |
CKMT2E7 |
tgggttggaacttgttcacctggcact |
ccatttagctcagtctgggcatgctgtcaa |
CKMT2E8 |
gcaatcaagctagggaggtaatgggaat |
tggaagttaccatagtgtcttctttca |
CKMT2E9 |
tctctggtgttagggatgtgctgaatg |
tgggcagtgatgttttcttcctccaca |
CKMT2E10 |
atgtcttttgtcagtaaacgcaccaat |
tgtgaaaacggctaggctgtttgcttt |
CKMT2E11 |
tggcaagttctcatttatcacagtgctgttcc |
gaggtgtgtgtaagttgtggcaaaatca |
CKMT1 |
|
|
CKMT1AE1F |
tctggattaaacagcattagggagaca |
tgcctctgcttctcatgatcccccactt |
CKMT1AE2F |
ggagccccacccctggagactgc |
cccggattctcacttctacccgcacct |
CKMT1AE3F |
aaagccagacatggccaactggacagc |
ggggctcagagtcttgggggaaagtga |
CKMT1AE4F |
gaaatcaatgcacaattgaagtct |
aggaaataaggtgaaggggttggagat |
CKMT1AE5F |
aatgcctaaggaaggtctctcctttcc |
tgggcctttcagagcccatgtctcatc |
CKMT1AE6F |
ggccagatgagacatgggctctgaaagg |
ggcaaggcaagaattgggagagatagagg |
CKMT1AE7F |
cctcttttgtcttcatgccctcataaatgc |
tggttttcccagtatggtcccagctggttt |
CKMT1AE8F |
tgtgggtctagctaagggagggtccag |
aagggtccactccctccccatcca |
CKMT1AE9F |
gctctgagcaggttcagggctctttcagg |
catcccggaggctcactcactgccata |
CKMT1AE10F |
atgagcaggcaagtcagtcagtgataa |
aatcccactgcctcttttcatttc |
PCR |
|
|
β2-MG |
ctcgcgctactctctctttctg |
gcttacatgtctcgatcccactt |
CKMT2-RNA |
ggccagtatcttttctaagttgctaactgg |
ttactttttgccaaactgaggcagagg |
Table 1. Primer Sequences Used for Sequencing, PCR and RFLP.
Results
A total of 20 AMD individuals were screened by sequencing for mutations in the CKMT1A and CKMT1B genes. Using flanking primers, all 10 exons for CKMT1A and CKMT1B were amplified and sequenced. Additionally, 500 bps of sequence upstream of the transcription start site of the CKMT1A and CKMT1B on human chromosome 15q15.3 were also screened for base variations. The coding regions for CKMT1A and CKMT1B were identical except for 4 base variations. During sequence analysis, these variations were used to distinguish the two genes. No new base variations were observed in the CKMT1A and CKMT1B genes (data not shown).
We then screened in 71 AMD patients all 11 exons of the sarcomeric CKMT2 gene located on chromosome 5q13.3. In exon 4, we found a putative termination mutation heterozygous NM_001825.2 c.274C>CT (92Q>X) in one individual, patient AMD-02-141 (Figure 2). In addition, we also found 4 other DNA variations in the CKMT2 gene in other AMD individuals. One of these 4 changes is a novel variation - 498G>GA (exon 1; Figure 3) found in patients AMD-02-30 and 02-44, while the others are known changes - 24191G>GA (rs2270823, exon 7), 33263 T>TC (rs34054011, exon 11), and 33440 G>GA (rs545, exon 11). The patient AMD-02-141 has an only son who was examined by a retinal specialist (DSB) and found to be phenotypically normal (age 55) with no signs of AMD. We then designed a restriction fragment length polymorphism (RFLP) assay as a means to rapidly and efficiently identify the truncating mutation c.274C>CT in the CKMT2 gene. This assay was used to screen the proband and his son as well as 72 additional individuals with AMD. We determined that the son did not inherited the mutation c.274C>CT (92Q>X) (Figure 4) and we were not able to detect the 274C>CT mutation in any of the 72 additional AMD patients (Table 2).
Exon |
1 |
4 |
7 |
11 |
11 |
11 |
4 |
SNP |
498 G>GA |
274 C>CT |
24191 G>GA |
33263 T>TC |
33440 G>GA |
C>A |
G>T |
rs |
Novel |
Novel |
rs2270823 |
rs34054011 |
rs545 |
rs15497 |
rs1063543 |
AMD: Sequencing |
|
|
|
|
|
|
| |
GG: 67/71 |
CC: 70/71 |
GG: 50/68 |
TT: 64/66 |
GG: 42/66 |
CC: 71/71 |
GG: 71/71 |
| |
GA: 04-71 |
CT: 1/71 |
GA: 15/68 |
TC: 2/66 |
GA: 18/66 |
CA: 0/71 |
GT: 0/71 |
| |
AA: 0/71 |
TT: 071 |
AA: 03/68 |
TT: 0/66 |
AA: 6/66 |
AA: 0/71 |
TT: 0/71 |
AMD: RFLP |
N/A |
CC: 72/72 |
N/A |
N/A |
N/A |
N/A |
N/A |
| |
|
CT: 0/72 |
|
|
|
|
|
| |
|
TT: 0/72 |
|
|
|
|
|
Gene Coordinates hg19:chr5:NC_000005.10 |
81233356 |
81252816 |
81257052 |
81266124 |
81266301 |
81266178 |
81252762 |
Amino Acid Change |
5' UTR |
92 Q>X |
Intron |
Intron |
3' UTR |
394 L>M |
74 A>S |
2021 Copyright OAT. All rights reserv
Table 2. CKMT2 SNP Results.
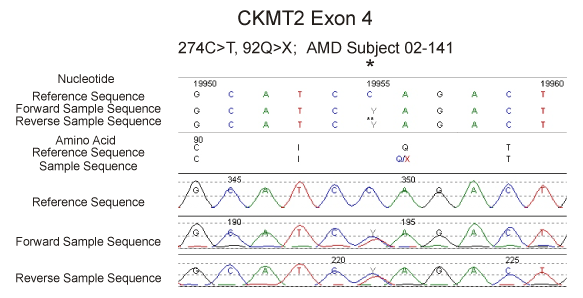
Figure 2. Sequencing of the CKMT2 gene showing the 274C>T mutation at the hgy19:chr5:81,252,816 position in patient AMD-02-141.
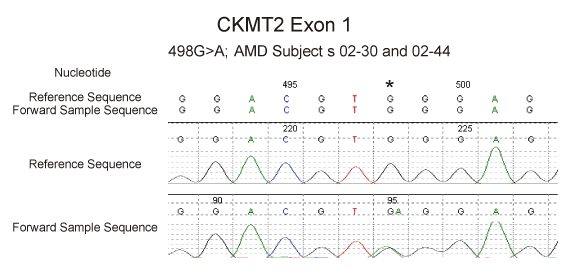
Figure 3. Sequencing of the CKMT2 gene showing the novel 498G>GA variant at position hg19:chr5:81,233,356 in two AMD patients.
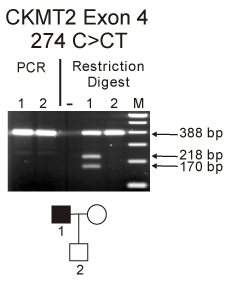
Figure 4. Upper panel: PCR amplification of the CKMT2 in the exon 4 region from proband patient AMD-02-141 (lane 1) and his son (lane2) showed a 388 bp product. After enzyme digestion with Xba1 the proband-AMD-02-141 showed two lower bands (218bp and 170bp) indicating the presence of the 274 C>CT SNP while the son did not have the change (lane 2). Lower panel: Pedigree for proband AMD-02-141. M, marker.
Although there was no information on the expression of the different CKMT2 transcripts in the retina, we wanted to determine if this gene was expressed within the human retinal RPE cells which are damaged early in AMD. The RNA was extracted from ARPE-19 cells and RT-PCR was performed. Figure 5 shows that the b2MG gene (334bp), representing nuclear DNA, was present in the ARPE-19 cells. Then RT-PCR was performed using primers for the CKMT2 gene and showed a product of the desired size (1258bp), demonstrating that the CKMT2 was expressed in human RPE cells. Interestingly, there was no CKMT2 gene expression from lymphocytes samples isolated from subject 02-141 and his son (data not shown). This was not totally unexpected since CKMT2 is usually associated with muscle cells and not blood cells.
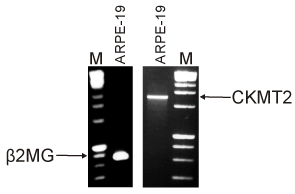
Figure 5. Gene expression of CKMT2 was identified in human ARPE-19 cells. RT-PCR amplification of RNA isolated from human ARPE-19 cells showed expression of both the β2MG (334bp) and CKMT2 (1258bp) genes. M, marker.
In the CKMT2 gene there are 19 non-synonymous SNPs that can cause amino acid changes as reported by NCBI Reference Assembly. Examination of the sequence patterns in the 71 AMD samples showed no mutations in these non-synonymous SNPs indicting that the CKMT2 gene is highly conserved.
Discussion
Recently it has been shown that AMD retinas have significant mitochondrial abnormalities including mtDNA damage [1-5]. In addition, studies on the mtDNA ancestral variants show that Northern European mtDNA haplogroups (J, T, and U) are associated with increased incidence of AMD [7,8,11-17]. Mutations associated with mitochondria are often associated with disorders of multiple systems. Tissues that have high energy requirements are susceptible to damage if mitochondrial dysfunction occurs due to defects in the mtDNA (i.e., MELAS, Leigh Syndrome, Leber Hereditary Optic Neuropathy, Kerns-Sayre Syndrome or Chronic Progressive External Ophthalmoplegia) or within the nuclear genes that are associated with mitochondrial function. Common clinical features associated with mitochondrial disorders include exercise intolerance, cardiac dysrhythmias, pigmentary retinopathy, sensorineural deafness, axonal neuropathy and endocrine abnormalities [18]. Jones and coworkers reported the A3243G mtDNA mutation in a patient with early age related maculopathy, hearing loss, hypertension, ischemic heart disease, and asthma [19]. A clinical phenotype of macular pattern dystrophy, deafness, and diabetes has been reported in subjects with a mtDNA point mutation at A3243G, which is located within the mitochondrial tRNA(Leu) gene [20,21]. Individuals with the novel MT-CYB gene mutation (A15579G), affecting complex III activity, exhibit a phenotype involving severe exercise intolerance, deafness, mental retardation, retinitis pigmentosa, cataract, growth retardation and epilepsy [22]. Therefore, mutations within the mtDNA are associated with characteristic patterns of symptoms.
In the present study, we identified two novel SNP variants in the nuclear DNA CKMT2 gene. One is a putative termination mutation (NM_001825.2 c.274C>T; 92Q>X, Exon 4) in an individual with AMD, exercise fatigue, atrial fibrillation, and deafness. Upon investigation, it is the first report of this mutation of the hg19:chr5:80548635 position in the CKMT2 gene, indicating that it is a highly conserved gene throughout mammals. The second previously unreported SNP variant (NM_001825.2.-498G>GA; 5’UTR, Exon 1) was found in 4 out of 71 subjects. The CKMT2 coordinates are at the 5q13.3 locus, located close to the defect causing Ushers Syndrome type 2C that includes deafness and retinitis pigmentosa [23].
Recent evidence demonstrates a relationship between mitochondria and the nuclear encoded CKMT2 gene. Human retinal cells lacking mtDNA (Rho0) have significantly upregulated expression of the CKMT2 gene [9]. While the CKMT2 gene has not been linked to a recognized mitochondrial disease, it has been associated with increased severity and resistance to anti-thyroid drug treatments for endocrinology patients with T3-predominant Graves’ thyroid disease [24]. CKMT2 is a marker for myogenic differentiation that can be upregulated by a novel actin-binding protein called Striated Muscle Activator of Rho Signaling (STARS) [25,26]. Recent studies show that CKMT2 and CKMT1A/B are exclusively produced in the highly metabolically active human brown fat but lacking in white adipose tissues [27]. The CKMTs utilize ATP to convert creatine (Cr) to phospho-creatine (p-Cr). The flow of metabolites is regulated by the mitochondrial creatine kinases, which structurally form complexes (mitochondrial interactosomes) with the mitochondrial voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane and the ADP/ATP-translocase in the inner mitochondrial membrane. In this scenario, it becomes obvious that diminished levels or mutations of the CKMT2 gene would have significant effects on energy homeostasis, production of reactive oxygen species and tissue longevity, likely leading to tissue dysfunction and diseases.
The earliest pathological changes in AMD are found in the RPE located beneath the neuro-retina. Our studies show CKMT2 expression in cultured human RPE cells, which is in agreement with other studies [9]. However to our knowledge, this the first reported mutation within the CKMT2 gene. The retina is among the most metabolically active tissues in the body and a mutation causing the production of a truncated form of CKMT2 could result in diminished efficiency for cells, increased levels of apoptosis and RPE cell death, which are common features in AMD. Since mitochondrial creatine kinase is such an important protein for energy production, the CKMT2 mutation would have serious consequences for all high energy requiring tissues and may potentially result in the multiple phenotypes described in our patient. Therefore, our data suggest that patients with the clinical picture of AMD plus deafness, exercise intolerance and/or cardiac dysarrhythmia should be screened not only for the mitochondrial DNA defect A3243G but also for the truncating mutation 274C>T in the CKMT2 gene.
Acknowledgments
We thank the individuals who donated blood samples to be used in this study and the research coordinators who worked on the study. This research was supported by the Discovery Eye Foundation, Lincy Foundation, Beckman Initiative for Macular Research, Henry Guenther Foundation, Polly and Michael Smith and non-restricted grant from Research to Prevent Blindness Foundation.
Competing interests
The authors declare that they have no competing interests. This research was conducted in accordance with institutional review board approved procedures.
Author’s contributions
MCK and NU helped design the study, interpret data, perform statistical analyses and write the manuscript. ABN and DSB performed clinical examinations on the patients and provided blood samples. NU designed the primers. SRA, DH, MM, and JL performed PCR and restriction enzyme digestion. SRA conducted sequencing analysis and figure generation. MC performed all tissue culture experiments.
References
- Kenney MC, Atilano SR, Boyer D, Chwa M, Chak G, et al. (2010) Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest Ophthalmol Vis Sci 51: 4289-4297. [Crossref]
- Nag TC, Wadhwa S, Chaudhury S (2006) The occurrence of cone inclusions in the ageing human retina and their possible effect upon vision: an electron microscope study. Brain Res Bull 71: 224-232. [Crossref]
- Bravo-Nuevo A, Williams N, Geller S, Stone J (2003) Mitochondrial deletions in normal and degenerating rat retina. Adv Exp Med Biol 533: 241-248. [Crossref]
- iang FQ, Godley BF (2003) Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res 76: 397-403. [Crossref]
- Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA (2008) Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci 49: 2848-2855. [Crossref]
- Canter JA, Olson LM, Spencer K, Schnetz-Boutaud N, Anderson B, et al. (2008) Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE 3: e2091. [Crossref]
- SanGiovanni JP, Arking DE, Iyengar SK, Elashoff M, Clemons TE, et al. (2009) Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS One 4: e5508. [Crossref]
- Jones MM, Manwaring N, Wang JJ, Rochtchina E, Mitchell P, et al. (2007) Mitochondrial DNA haplogroups and age-related maculopathy. Arch Ophthalmol 125: 1235-1240. [Crossref]
- Miceli MV, Jazwinski SM (2005) Common and cell type-specific responses of human cells to mitochondrial dysfunction. Exp Cell Res 302: 270-280. [Crossref]
- Seddon JM, Sharma S, Adelman RA (2006) Evaluation of the clinical age-related maculopathy staging system. Ophthalmology 113: 260-266. [Crossref]
- van der Walt JM, Nicodemus KK, Martin ER, Scott WK, Nance MA, et al. (2003) Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet 72: 804-811. [Crossref]
- van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, et al. (2004) Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett 365: 28-32. [Crossref]
- Hassani-Kumleh H, Houshmand M, Panahi MS, Riazi GH, Sanati MH, et al. (2006) Mitochondrial D-loop variation in Persian multiple sclerosis patients: K and A haplogroups as a risk factor!! Cell Mol Neurobiol 26: 119-125. [Crossref]
- Dunaief JL, Dentchev T, Ying GS, Milam AH (2002) The role of apoptosis in age-related macular degeneration. Arch Ophthalmol 120: 1435-1442. [Crossref]
- Huerta C, Castro MG, Coto E, Blázquez M, Ribacoba R, et al. (2005) Mitochondrial DNA polymorphisms and risk of Parkinson's disease in Spanish population. J Neurol Sci 236: 49-54. [Crossref]
- Udar N, Atilano SR, Memarzadeh M, Boyer D, Chwa M, et al. (2009) Mitochondrial DNA haplogroups associated with Age-related macular degeneration. Invest Ophthalmol Vis Sci 50: 2966-2974. [Crossref]
- Mueller EE, Schaier E, Brunner SM, Eder W, Mayr JA, et al. (2012) Mitochondrial haplogroups and control region polymorphisms in age-related macular degeneration: a case-control study. PLoS ONE 7: e30874. [Crossref]
- Edmond JC (2009) Mitochondrial disorders. Int Ophthalmol Clin 49: 27-33. [Crossref]
- Jones M, Mitchell P, Wang JJ, Sue C (2004) MELAS A3243G mitochondrial DNA mutation and age related maculopathy. Am J Ophthalmol 138: 1051-1053. [Crossref]
- Bonte CA, Matthijs GL, Cassiman JJ, Leys AM (1997) Macular pattern dystrophy in patients with deafness and diabetes. Retina 17: 216-221. [Crossref]
- Michaelides M, Jenkins SA, Bamiou DE, Sweeney MG, Davis MB, et al. (2008) Macular dystrophy associated with the A3243G mitochondrial DNA mutation. Distinct retinal and associated features, disease variability, and characterization of asymptomatic family members. Arch Ophthalmol 126: 320-328. [Crossref]
- Wibrand F, Ravn K, Schwartz M, Rosenberg T, Horn N, et al. (2001) Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene. Ann Neurol 50: 540-543. [Crossref]
- Yan D, Liu XZ (2010) Genetics and pathological mechanisms of Usher syndrome. J Hum Genet 55: 327-335. [Crossref]
- Matsumoto C, Ito M, Yamada H, Yamakawa N, Yoshida H, et al. (2013) Genes that characterize T3-predominant Graves' thyroid tissues. Eur J Endocrinol 168: 137-144. [Crossref]
- Wallace MA, Russell AP (2013) Striated muscle activator of Rho signaling is required for myotube survival but does not influence basal protein synthesis or degradation. Am J Physiol Cell Physiol 305: C414-426. [Crossref]
- Wallace MA, Della Gatta PA, Ahmad Mir B, Kowalski GM, Kloehn J, et al. (2016) Overexpression of Striated Muscle Activator of Rho Signaling (STARS) Increases C2C12 Skeletal Muscle Cell Differentiation. Frontiers in Physiology 7: 7. [Crossref]
- Muller S, Balaz M, Stefanicka P, Varga L, Amri EZ, et al. (2016) Proteomic Analysis of Human Brown Adipose Tissue Reveals Utilization of Coupled and Uncoupled Energy Expenditure Pathways. Scientific Reports 6: 30030. [Crossref]