Background: Craniofacial fibrous dysplasia, is a rare disease with aesthetic and neurological alterations, sometimes requiring complex surgical treatment. The aim of this study is to describe the clinical course and treatment in fibrous dysplasia patients at one of the highest-demanding cancer centres in Latin America.
Methods: Retrospective case series study from 1984 to 20019. Demographic, epidemiological, and clinical variables were analysed. Two representative cases were described.
Results: In a thirty-five years period, nine patients were included. The mean age at diagnosis was 25.3 9.34 years, 55.5% were female. The most common affected was the frontal bone in 88.8%, skull base was involved in 77.7%. The mean follow-up time was 17.5 (±11.43) years, trigeminal nerve was the most commonly involved. Karnofsky of 95.5+7.2. At the end of the study, Karnofsky score was on average 91.1+13.6.
Conclusion: There is a diverse clinical course, sometimes with high morbidity and many patients require multiple surgical interventions during the evolution of this condition. Close monitoring allows appropriate behaviours to be taken to avoid permanent neurological deficits or unwanted cosmetic defects by patients.
fibrous dysplasia, Skull, Craniofacial, Skull tumour
Fibrous dysplasia (FD), was described by Lichtenstein in 1938 [1], as a progressive lesion in which normal bone elements are replaced by fibrous tissue, associated with McCune-Albright syndrome [2-4]. In most cases, it behaves like a benign disease with a slow progression [5]. It is a rare disease in the general population with an incidence of 1: 5000 to 1: 10,000/ year [6], however, accounts for approximately 10% of all bone tumours [7]. It is a benign bone lesion where there is a turnover from normal bony components to premature stromal tissue as a result of GNAS mutation [8,9], with the potential to cause aesthetic and functional alterations, particularly in the craniofacial bones [10]. The skull base is the most commonly affected site on the craniofacial skeleton [11], and may present as craniomaxillofacial, orbital, fronto-orbital, cranio-orbital, or complex FD [12-15]. GNAS gene somatic mutations, located on the long arm of chromosome 20 (20q13.2) are responsible for FD [16,17]. The mutations alter the α subunit of the Gs protein with associated excessive activation of the AMPC pathway, leading to cell proliferation and poor differentiation [18-20]. Exchange of arginine by cysteine or histidine on the 201 position in the Gsα protein is also found on FD associated with McCune-Albright syndrome [21,22].
FD can present as Two distinct forms: 1) monostotic variant where a single bone is involved and comprises 70% of cases and 2) polyostotic variant where multiple bones are involved, nonetheless, the latter is the form most commonly affecting the craniofacial skeleton [16].
Deformity and swelling are the most characteristic features in FD [17]. Hearing loss, either by external auditory canal stenosis (conductive) or by affecting the otic capsule (sensorineural) due to temporal involvement [18]; sinusitis as a result of frontal, sphenoid, nasoethmoid and maxilar involvement [17], skull base and orbital FD with it subsequent dystopia, diplopia, diminished visual acuity, proptosis, headaches and facial pain from trigeminal neuralgia are also frequent clinical findings [19-22]. Visual loss due to FD is a major concern [23], as it greatly modifies the quality of life [6], around 95% of patients present with no visual impairment [24], and if it is present, endoscopic optic nerve decompression can be performed [25].
This study aims to describe the clinical course and treatment in FD patients at one of the highest-demanding cancer centres in Latin America.
We retrospectively studied records of patients from 1985 to 2019 in the Neurosciences unit at the National Cancer Institute of Mexico (INCan, Mexico City). The study was approved by the institutional review board. Inclusion criteria were 1) patients diagnosed with Craniofacial FD of both genders, 2) 18 years of age or more and 3) complete clinical and radiographic data. Patients with incomplete data on their clinical records were excluded. Demographic, epidemiological, and clinical variables were analysed. Finally, two complex representative cases are summarized
Statistical analysis was done using SPSS 26.0 (IBMTM) statistical software. Descriptive statistics was reported in means, ± standard deviation (± SD), or percentage.
In a thirty-five years period, nine patients were included (Table 1). The mean age at diagnosis was 25.3 9.34 years, 55.5% were female. The affected bones were as follows: frontal bone 88.8%, ethmoid, and maxilla in 66.6%, sphenoid in 55.5%, parietal in 22.2%, and occipital in 11.1% of patients. The skull base was involved in 77.7% and the cranial vault in 22.2% of patients. The alkaline phosphatase was found elevated in only one case (11.1%).
The mean follow-up time was 17.5 (±11.43) years. Patients presented at the time of diagnosis with a Karnofsky of 95.5+7.2. At the end of the study, Karnofsky’s score was on average 91.1 +13.6.
Table 1. Mean
Patient |
Age at the time of study |
Gender |
Year |
Type |
Facial deformity |
First Symptom |
CN affection |
KPS at diagnoses / Current FD |
VA |
1 |
30 |
FEMALE |
2004 |
MONOSTOTIC |
YES |
DYSMORPHISM |
NO |
100/100 |
20/20 |
2 |
48 |
FEMALE |
2007 |
MONOSTOTIC |
YES |
DYSMORPHISM |
NO |
100/100 |
20/20 |
3 |
41 |
FEMALE |
2003 |
MONOSTOTIC |
YES |
DYSMORPHISM |
NO |
100/100 |
20/20 |
4 |
33 |
FEMALE |
2006 |
MONOSTOTIC |
YES |
NASAL CONGESTION |
RIGHT II |
100/100 |
20/25 |
5 |
42 |
MALE |
2010 |
MONOSTOTIC |
YES |
HEADACHE |
RIGHT V1 V2 |
80/60 |
20/20 |
6 |
41 |
MALE |
2012 |
POLYOSTOTIC |
NOT |
HEADACHE |
RIGHT III |
90/90 |
20/20 |
7 |
44 |
MALE |
2012 |
MONOSTOTIC |
YES |
DYSMORPHISM |
NO |
90/90 |
20/30 |
|
|
|
|
|
|
|
|
|
|
8 |
52 |
FEMALE |
1987 |
MONOSTOTIC |
YES |
DYSMORPHISM |
RIGHT V1 V2 |
100/80 |
20/70 |
9 |
32 |
MALE |
2019 |
POLYOSTOTIC |
YES |
DYSMORPHISM |
NO |
100/100 |
20/20 |
The monostotic variant was the most frequent, presented in 77.75% of the cases. No clinical evidence of Jaffe-Lichtenstein or McCune Albright Syndrome was evident, although we lack laboratory confirmation. Only one patient had concomitant neoplasia, presenting with a salivary glands cystic adenoid carcinoma. The most common sign was a facial or cranial deformity in up to 88.8%. Other symptoms were headache in the 22.2% and nasal obstruction in 11.1%. Cranial nerve palsy or neurological deficit was not reported as the main cause of complaint. At the physical evaluation, we found the following: exophthalmos in 55.5% and cranial nerve involvement in 44.4% (optic nerve in 11.1%, oculomotor in 11.1%, and trigeminal nerve in 22.2%. Less frequent findings were xerosis, limitation to oral occlusion, and epistaxis.
Headache was presented in 55.5% of cases that mostly improved with the use of analgesics, antineuritics, and bisphosphonates (ibandronic or zolendronic acid). 44.4% were offered surgical management, 33.3% agreed with a surgical strategy.
Case 1
19 years-old female previously healthy, presented to neurosurgery outpatient clinic with complaints beginning at 10 years of age. She started with a slow and progressive increase in volume on the forehead. During this time the lesion gained size and was followed by frontal oppressive headache and epistaxis. Physical examination showed hypertelorism, a disfiguring left frontal lesion, hypoesthesia in right V1 territory, and partial obstruction of the choanae. Imaging studies revealed an expansive bone lesion in the frontal, sphenoid, and ethmoidal sinus and invasion of the orbital medial wall with compression and displacement of the eye (Figure 1). Due to its compressive effect, we preferred a transmaxilar approach. The patient had an uneventful postoperative period and had a follow-up in an outpatient setting. However, after 46 months, the symptoms recurred. After proper clinical and imaging evaluation, we performed a frontoorbitary craniectomy with the remodelling of the adjacent tissue, and at the same time a cranioplasty with methyl methacrylate. Frontal sinusitis presented one year after the second surgical event, requiring cranialization of the frontal sinus. Three years later the cranioplasty plate had to be removed due to exposure, therefore we undertook remodelling of the cranioplasty plate and primary wound closure.
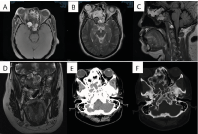
Figure 1. Images of case 1 are presented, corresponding to patient number 8 in table 2. (A) Axial MRI sequence T1 and (B) T2, where can be observed the displacement of the soft structures of the orbit with protrusion of the right ocular globe, in the midsagittal section (C) lesion with cystic appearance that involves sphenoid and clivus, with a displacement of the frontal parenchyma. Coronal section T2 (D) where it invades the nasal cavity and sinuses. In computed tomography of the skull base, (E,F) can be observed the extension of the lesion
Case 2
A 44-year-old male, with a previous FD history, since he was 10 years old, began with a frontal and left hemicrania oppressive headache and enlargement of the periorbital region. Initially, he had conservative management for osteoma in another institution and later referred to our institution at age 37 due to evident facial deformity. After clinical and imaging studies, FD was diagnosed (Figure 2). A combined craniofacial approach was planned for resection of lesion in the frontal bone and lesser wing of the sphenoid bone, including an optic nerve decompression, plus placement of a prefabricated methyl methacrylate plate, taking as a template, the presurgical images (Figure 3).
During the first year of follow-up, the patient reported intense headaches. The patient mentioned having diplopia, and ocular misalignment was evident (Figure 3), the reason to undergo orbital reconstruction with iliac crest graft. During his subsequent medical visits, he referred to persist with diplopia.
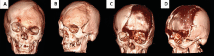
Figure 2. Clinical case 2, patient number 7 in table 1. CT with three-dimensional reconstruction, pre-surgical, anteroposterior view, (A) anterior oblique view (B), where the bone lesion was observed and post-surgical study, cranioplasty, anterior view (C), and anterior oblique with methyl methacrylate material by stereolithography (D)
Discussion
FD is a condition that can also be part of the Jaffe-Lichtenstein syndrome with polyostotic FD [26,27], or associated with endocrine disarrangements, multiple bone affections, skin darkening, and cutaneous café-au-lait spots, known as McCune-Albright syndrome [28]. A clear polyostotic predominance is reported in the literature [2,29], however, in this case series, the monostotic variant is the most frequently associated with the craniofacial region, and only one case of the polyostotic variant was observed (Figure 4). Clinical evidence of cutaneous or endocrine manifestation associated with FD as described by several authors was not documented in any of the patients described before [29-31]. In this study, FD behaves as previously reported, presenting as a massive lesion with insidious growth, with symptoms arising from compression or displacement of contiguous structures, such as the brain, optic nerve, eye, airway, cranial nerves, and middle ear [16]. Headache or dysmorphism were present in most of the patients, other symptoms were deformity and facial asymmetry, visual defects, hearing impairment, nasal congestion or nasal obstruction, facial pain, paraesthesia, and oral malocclusion as a result of the affected bone site. Nonetheless, patients can be frequently asymptomatic [30-32]. Usually, the lesions appear during the first decade of life [32], with faster growth in children and adolescents due to cortical bone expansion and have an association with other pathologies such as aneurysmal bone cysts, mucoceles, or malignant transformation to osteosarcoma, however, these associations represent less than 1%.
Some cardinal symptoms require urgent attention such as rapidly evolving facial discrepancy, acute onset of pain, acute loss of hearing, airway obstruction, or new-onset paraesthesia [33].
Sometimes aggressive surgical resection is recommended to avoid these complications [16]. However, it has recently been shown that aggressive behaviour of the disease, that is, rapid expansion is the exception for that choice of treatment and conservative management is more prudent [4,34,35], as in most of our patients.
In the National Cancer Institute, the mean age of diagnosis was 24.5 years, yet many patients have an onset at a very earlier age, which means there is a delay in diagnosis and treatment due to the insidious and sometimes vague symptoms, worries of the patients or a low clinical suspicion when evaluating the patients at first contact.
90% of FD lesions, regardless of site, are present before 15 years old, especially in the craniofacial region; moreover, the age of 10, marks the limit for the onset of the lesion in the craniofacial region [3].
The degree of facial deformity is variable and depends upon the affected bone and the growing speed; it can cause a wide number of symptoms ranging from facial asymmetry to more severe conditions such as vertical dystopia and proptosis [36]. These features are classified in Table 2.
Table 2. Feature Classification
Grade |
Feature |
Tracing |
Treatment |
1 |
Without growth (quiescent) |
Require observation and control with annual monitoring |
Annual clinical follow-up with serial photographs and craniofacial tomography, and the surgical treatment is reduced to the aesthetic field trying to eliminate the complete lesion with facial reconstruction and follow-up to determine that there is no recurrence. |
2 |
Slow growth (not aggressive). |
Wait until the lesion is inactive and the patient reaches bone maturity to offer surgical treatment. |
Surgery in case of affection of the psychosocial development due to facial deformity or functional alteration of some CN. Informing about the possibility of recidivism, which may require more interventions according to the evolution. |
3 |
Rapid growth (aggressive), characterized by pain, paresthesia, pathological fractures, associated with other pathologies or with malignant transformation. |
It is a priority to perform a biopsy |
Complement the treatment with the en bloc resection of the lesion.
Malignant transformation of Fibrous Dysplasia has been reported in less than 1% of cases. |
Cranial nerve compression is found in 44.4% of patients, and it is determined by the site of dysplasia; this high percentage correlates with the most frequent bones affected. FD with skull base involvement is characterized by dystopia, hypertelorism, and proptosis through frontal, sphenoid, and ethmoid bones. Skull base involvement can also originate optic neuropathy, strabismus, oral malocclusion, obstruction of the nasolacrimal duct, and trigeminal neuralgia [13].
The sphenoid sinus is the most frequently affected, followed by the ethmoidal and maxillary sinuses [37], this fact was not observed in the cases described since paranasal sinuses were equally affected.
Endoscopic approaches are considered as first-line treatment for lesions located in the nasal cavity or paranasal sinuses [38], our treatment was based upon broad approaches to perform the maximum possible resection of the lesion, with the intent to perform a reconstruction through stereolithography with methylmethacrylate in the same procedure (Figure 3).
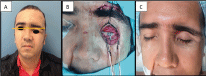
Figure 3. Image of clinical case 2, patient number 7 in table 1. Patient after first surgery. Diplopia and ocular height difference were noted (A), which was subjected to a second surgical correction event, placing methyl methacrylate plate (B), in image C, we observed a symmetrical correction of ocular height
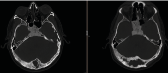
Figure 4. CT with bone window of patient number 6 in table 2. Fired glass images are shown as characteristic of FD. Multiple areas were involved in this patient, polyostotic FD was diagnosed
The most common complications in surgically treated patients are sinusitis and exposure of the methyl methacrylate material, with the need for a second surgical procedure.
Occasionally the diagnosis is made incidentally during routine imaging such as X-rays or CT scan or when facial or cranial asymmetry is noted [30,31]. One patient did not have a facial deformity, however, during imaging studies for salivary gland cancer, osteolytic lesions were reported on frontal and parietal bone, for which we performed biopsy sampling discarding metastasis and diagnosing FD. FD has a radiological aspect of radiolucent lithic lesions, with a homogeneous appearance of frosted glass and indefinite borders [39] (Figure 5). Occasionally, it may reveal as sclerotic lesions with or without lytic lesions [4]. Thus, FD can present as three main radiological patterns: 1) pagetoid (56%), 2) sclerotic (23%) and 3) cystic (21%) [2]. Recently it has been described the use of bone scan with 99Tc-MDP [30, 31], although it is not widely available due to high costs. Currently, CT scans are used to not only further understand the morphology, but to determine the extent and plan the surgical approach. This is done by obtaining images from the vertex to the thyroid region and using CT images no more than 3.75 mm thick [39].
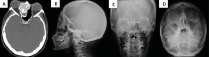
Figure 5. Images of the case of patient number 4 in table 2. Skull CT (A), X-ray of skull lateral projection (B), Anteroposterior (C), and Waters position (D), where we observed the superposition of the bones of the skull base on radiographs and makes it more difficult to identify bone abnormalities in inexperienced eyes, compared with the sensitivity of CT
Simple radiographs are no longer recommended for the diagnosis of cranial or facial lesions due to overlapping skull base bones [39]. MRI is useful to evaluate soft tissue, and fibrous components and to assess compression of adjacent structures. The MRI imaging features of FD are variable, hypointense, or isointense in T2 with small regions of T2 hyperintensity that can represent cystic regions with variable enhancement patterns [40] (Figure 1).
Clinical and imaging evaluations were performed annually; the follow-up time of our patients was very diverse since being a slow progression disease, allows long periods of active or inactive illness, having an average of 17.5 years of follow-up for our patients.
Although biopsy is the gold standard to confirm the diagnosis, lack of symptoms and inaccessible surgical location may prompt using imaging studies for diagnosis [4]. Histology is characterized by immature tissue composed of irregular anastomotic trabeculae of partially calcified osteoid tissue, abnormal fibrous connective tissue, absence of mature cortical tissue, coated by a layer of activated osteoblasts [41].
When ruling out other diagnoses is needed, immunohistochemical analysis with MDM2 and CDK4 can help distinguish FD from a malignancy since neoplasms will often express MDM2 or CDK4 while FD will not [42].
The clinical behaviour dictates the management since the histological pattern lacks prognostic value and no biomarkers are currently available, hence it is imperative to grant close monitoring to every patient, especially the pediatric population. FD is difficult to diagnose and to successfully treat [43], because of low clinical suspicion which masks the actual incidence. Treatment is burdensome since cranial nerves and eloquent areas may be affected.
Bland, et al. describes a gradual venous compression leading to retinal ischemia and visual loss [20], besides the very common compression of optic and oculomotor nerves; this makes the neuroophthalmological evaluation fundamental, focusing on assessing optic neuropathy and must include visual acuity, visual field examination, contrast sensitivity, colour vision, and fundus examination, pupillary reflexes, eye movements, monitoring proptosis with ophthalmometry, eyelid closure, hypertelorism and tear duct [36]. Nonetheless, only a few patients become blind even in the presence of compression evidence from CT images [4,44], consequently, prophylactic decompression is not indicated [4]. Early identification of acute deficit may prompt early surgical treatment for decompression [6,34], however, the timing for optic nerve decompression is not well defined [20], since no medical therapy has proven efficacy to stop or turn around the progression [45-47]. In our series only one patient presented with optical compression and a transcranial decompressive procedure was performed, however, DeKlotz and Ryu advocate for endoscopic endonasal procedures [25,40].
Up to 12% of patients with polyostotic FD have elevated levels of growth hormone, increasing 4.1 times the risk of optic neuropathy due to compression [44].
This series fails to show clinical features of abnormal growth hormone concentrations because not all patients had endocrine laboratory tests done at the initial evaluation nor during follow-up. Denosumab, a ligand RANK inhibitor, is a promising option [39], for fibrous dysplasia exhibiting an accelerated evolution, nonetheless, this clinical course occurs mainly in the paediatric age. Radiotherapy may increase the risk of bone tissue becoming malignant, consequently is not recommended.
It should be noted that although it is a disease that can potentially cause changes in patients lifestyle, their functional status and independence was not significantly affected as 55.5% persisted with a Karnofsky of 100 points from their diagnosis to their last evaluations, 33.3 % had a decrease to 80 - 90 points without affecting their independence and quality of life, only one patient (11.1%) had 60 points due to trigeminal neuralgia with limitation of daily activities, but, in the end, the condition remitted before being managed surgically.
There is no current report in Latin America of any case series of Craniofacial FD with this number of patients or during this time period, in which complex cases are presented.
FD is a relatively benign disease with slow progression and variable clinical course, the reason why it is sometimes minimized by some physicians, yet it can affect the quality of life due to disability or mostly due to social implications because of deformity, so treatment must be individualized and multidisciplinary.
Undoubtedly, the diagnosis and treatment are a challenge for the medical team because imaging studies are not highly specific for this condition and biopsy can be extremely invasive besides it is impossible to completely resect the lesion during surgical procedures. The immunological therapy with Denosumab improves the quality of life; and on the other hand, radiotherapy plays a contradictory role as a predisposing factor for malignant transformation. There is diverse clinical course, with high morbidity and many patients require multiple surgical interventions during the evolution of this condition. Close monitoring allows appropriate behaviours to be taken to avoid permanent neurological deficits or unwanted cosmetic defects by patients.
- Lichtenstein L, Jaffe HL (1942) Fibrous dysplasia of bone: a condition affecting one, several or many bones, the graver cases of which may present abnormal pigmentation of skin, premature sexual development, hyperthyroidism or still other extraskeletal ab- normalities. Arch Pathol 33: 777. [Crossref]
- McCune DJ (1936) Osteitis fibrosa cystica: the case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 52: 743-747.
- Albright F, Butler AM, Hampton AO, Smith P (1937) Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females: report of five cases. N Engl J Med 216: 727-746.
- Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, et al. (2002) Normal vision despite the narrowing of the optic canal in fibrous dysplasia. N Engl J Med 347: 1670-1676. [Crossref]
- Pai B, Ferdinand D (2013) Fibrous dysplasia causing safeguarding concerns. Arch Dis Child 98: 1003-1006.
- Amit M, Collins MT, FitzGibbon EJ, Butman JA, Fliss DM (2011) Surgery versus Watchful Waiting in Patients with Craniofacial Fibrous Dysplasia - a Meta-Analysis. PLoS ONE 6: e25179 [Crossref]
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, et al. (1991) Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 325: 1688-1695. [Crossref]
- Schwindinger WF, Francomano CA, Levine MA (1992) Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci U S A 89: 5152-5156. [Crossref]
- Edgerton MT, Persing JA, Jane JA (1985) The surgical treatment of fibrous dysplasia with emphasis on recent contributions from craniomaxillofacial surgery. Ann Surg 202: 459. [Crossref]
- Jackson IT, Hide TAH, Gomuwka PK (1982) Treatment of cranioorbital fibrous dysplasia. J Maxillo fac Surg 10: 138-141. [Crossref]
- Munro IR, Lauritzen CG (1985) Periorbital tumors, In Caronni E (Eds): Craniofacial Surgery. Boston MA, Little, Brown and Company pp: 329-344.
- Munro IR, Chin B, Chen YR (1981) Radical treatment for fronto-orbital fibrous dysplasia: The chain-link fence. Plast Reconstr Surg 67: 719-730. [Crossref]
- Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, et al. (2012) Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis 24: S2. [Crossref]
- Marie PJ, de Pollak C, Chanson P, Lomri A (1997) Increased proliferation of osteoblastic cells expressing the activating Gs a mutation in monostotic and polyostotic fibrous dysplasia. Am J Pathol 150: 1059-1069. [Crossref]
- Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, et al (1997) Fibrous dysplasia of bone in the McCune-Albright syndrome: Abnormalities in bone formation. Am J Pathol 151: 1587-1600. [Crossref]
- Ricalde P, Horswell BB (2001) Craniofacial fibrous dysplasia of the fronto-orbital region: a case series and literature review. J Oral Maxillofac Surg 59: 157- 167. [Crossref]
- Moore RT, Buncic JR, Munro IR (1985) Fibrous dysplasia of the orbit in childhood: Clinical features and management. Ophthalmology 92: 12-20. [Crossref]
- Katz BJ, Nerad JA (1998) Ophthalmic manifestations of fibrous dysplasia: A disease of children and adults. Ophthalmology 105: 2207-2215. [Crossref]
- Morrissey MD, Talbot JM, Schleuning AJ II: Fibrous dysplasia of the temporal bone: Reversal of sensorineural hearing loss after decompression of the internal auditory canal. Laryngoscope 107: 1336-1340. [Crossref]
- Bland LI, Marchese MJ, McDonald JV (1992) Acute monocular blindness secondary to fibrous dysplasia of the skull: A case report. Ann Ophthalmol 24: 263. [Crossref]
- Osguthorpe JD, Gudeman SK (1987) Orbital complications of fibrous dysplasia. Otolaryngol Head Neck Surg 97: 403. [Crossref]
- Liakos GM, Walker CB, Carruth JAS (1979) Ocular complications in craniofacial fibrous dysplasia. Br J Ophthalmol 63: 611-616. [Crossref]
- Sassin JF, Rosenberg RN (1968) Neurological complications of fibrous dysplasia of the skull. Arch Neurol 18: 363-369. [Crossref]
- Chen YR, Breidahl A, Chang CN (1997) Optic nerve decompression in fibrous dysplasia: indications, efficacy, and safety. Plast Reconstr Surg 99: 22-30. [Crossref]
- DeKlotz TR, Stefko ST, Fernandez-Miranda JC, Gardner PA, Snyderman CH, et al. (2017) Endoscopic Endonasal Optic Nerve Decompression for Fibrous Dysplasia. J Neurol Surg B Skull Base 78: 24-29. [Crossref]
- Faivre L, Nivelon-Chevallier A, Kottler ML, Robinet C, Khau Van Kien P, et al. (2001) Mazabraud syndrome in two patients: Clinical overlap with McCune-Albright syndrome. Am J Med Genet 99: 132-136. [Crossref]
- Zoccali C, Teori G, Prencipe U, Erba F (2009) Mazabraud's syndrome: A new case and review of the literature. Int Orthop 33: 605-610. [Crossref]
- Mishra N, Rout SK (2018) First two cases of craniomaxillofacial fibrous dysplasia from Nepal - case series. Clin Cosmet Investig Dent 10: 269-274. [Crossref]
- Lustig LR, Holliday MJ, McCarthy EF, Nager GT (2001) Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg 127: 1239-1247. [Crossref]
- Boyce AM, Florenzano P, de Castro LF, Collins MT (1993) Fibrous Dysplasia/McCune-Albright Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, et al. (Eds). GeneReviews. Seattle (WA). [Crossref]
- Robinson C, Collins MT, Boyce AM (2016) Fibrous Dysplasia/McCune-Albright Syndrome: Clinical and Translational Perspectives. Curr Osteoporos Rep 14: 178-186. [Crossref]
- Utriainen P, Valta H, Bjornsdottir S, Makitie O, Horemuzova E (2018) Polyostotic Fibrous Dysplasia With and Without McCune-Albright Syndrome- Clinical Features in a Nordic Pediatric Cohort. Front Endocrinol (Lausanne) 9: 96. [Crossref]
- Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, et al. (2007) Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res 22: 1468-1474. [Crossref]
- Diah E, Morris DE, Lo LJ, Chen YR (2007) Cyst degeneration in craniofacial fibrous dysplasia: clinical presentation and management. J Neurosurg 107: 504-508. [Crossref]
- Chen YR, Chang CN, Tan YC (2006) Craniofacial fibrous dysplasia: an update. Chang Gung Med J 29: 543-349. [Crossref]
- Pfeiffer J, Kayser G, Boedeker CC, Ridder GJ (2008) Posttraumatic reactive fibrous bone neoformation of the anterior skull base mimicking osteosarcoma. Skull Base 18: 345-351. [Crossref]
- Valentini V, Cassoni A, Terenzi V, Della Monaca M, Fadda MT, et al. (2017) Our experience in the surgical management of craniofacial fibrous dysplasia: what has changed in the last 10 years? Acta Otorhinolaryngol Ital 37: 436-443. [Crossref]
- Sciarretta V, Pasquini E, Frank G, Modugno GC, Cantaroni C, et al. (2006) Endoscopic treatment of benign tumors of the nose and paranasal sinuses: a report of 33 cases. Am J Rhinol 20: 64-71. [Crossref]
- Andreu-Arasa VC, Sung EK, Fujita A, Saito N, Sakai O (2019) Otosclerosis and Dysplasias of the Temporal Bone. Neuroimaging Clin N Am 29: 29-47. [Crossref]
- Ryu G, Al-Magribi AZ, Lee KE, Lee JJ, Kim SB, et al. (2020) Endoscopic optic nerve decompression for optic neuropathy in sinonasal fibro-osseous tumors. World Neurosurg 138: e260-e266. [Crossref]
- Jo VY, Fletcher CD (2014) WHO classification of soft tissue tumors: an update based on the 2013 (4th) edition. Pathology 46: 95-104. [Crossref]
- Dujardin F, Binh MB, Bouvier C, Gomez-Brouchet A, Larousserie F, et al. (2011) MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol 24: 624-637. [Crossref]
- Chang CY, Wu KG, Tiu CM, Hwang B (2002) Fibrous dysplasia of mandible with chronic osteomyelitis in a child: report of one case. Acta Paediatr Taiwan 43: 354-357. [Crossref]
- Cutler CM, Lee JS, Butman JA, FitzGibbon EJ, Kelly MH, et al. (2006) Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurgery 59: 1011-1017. [Crossref]
- Dumitrescu CE, Collins MT (2008) McCune-Albright syndrome. Orphanet J Rare Dis 3: 12. [Crossref]
- Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, et al. (2003) Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab 88: 4569-4575. [Crossref]
- DiMeglio LA (2007) Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev 4: 440-445. [Crossref]