The study of weed genomics is important for the effective management of weeds to enhance crop yield. A rapid, inexpensive, and high-quality DNA extraction is needed for genomic and other molecular studies. Here, we describe the protocols for DNA extraction from two different parts of the Echinochloa colona plant using modified cetyltrimethylammonium bromide (CTAB) and a commercial kit. In the study, it was observed that the DNA extracted from plant leaf tissues and dry seeds with a modified CTAB protocol was of good quality, with no contaminations of polysaccharides and essential oils. Quality of DNA extracted from dry seeds was comparable with that of plant leaves under both protocols. The extracted DNA from both plant parts was successfully amplified by PCR using the EPSPS microsatellite marker. Compared to the protocol of DNA extracted from leaf tissue, dry seeds will save time and other valuable resources. Moreover, the same protocols can be implemented for the extraction of high-quality DNA for molecular studies in other plant species where a large amount of polysaccharides, secondary metabolites and essential oils are present.
DNA extraction, PCR amplification, secondary metabolites, seeds, Echinochloa colona
The discovery of gene function in model plants has revolutionized the molecular genetics that can be applied in weed plants to understand their biology and effective management. The study of genomics in weeds will help in the identification of genes showing competitiveness for nutrients in crop fields [1]. Molecular techniques help in the identification of the gene responsible for rapid root and shoot development, early crop shoot emergence, identification of novel herbicide target sites, production of allelochemicals and resistance mechanisms. These techniques will dramatically affect the interaction between weed and crop and significantly reduce associated problems in-field. Weed genomics help in understanding the genetic composition of weed populations that change over time in response to weed management practices in agricultural crop fields.
High throughput DNA sequencing techniques help in the understanding of how and which gene (s) affect the fitness of weeds, their competitiveness and adaptation in different environments [1]. These techniques aid in the identification of genes contributing to weediness, herbicide resistance, weed reproductive characteristics, perennial growth habit, seed and vegetative structure, dormancy and weed plant morphological traits. Moreover, meagre genomic resources are available in weeds compared to field crops [2]. The implications of molecular techniques increase the potential for understanding of weed biology, herbicide mechanisms of action and resistance mechanisms. This will help in the identification of target sites of new herbicides and their development for grower use in management practices. For molecular biological studies, DNA extraction is the first step for many of the available downstream applications [3]. Successful extraction of DNA involves effective disruption of cells, denaturation of lipids, proteins and nucleoprotein complexes and inactivation of nucleases [4]. These precede the process of DNA pellet formation. The efficiency and success of molecular techniques mostly depend upon the quality of DNA extracted from genotypes [5]. In weeds and some cereals, the extraction of high-quality DNA is difficult due to the presence of proteins, polysaccharides, and other DNA polymerase inhibitors such as alkaloids, tannins, polyphenols etc. All these compounds affect the quality and quantity of isolated DNA and reduce the amplification of samples in polymerase chain reaction (PCR) [6]. For high-quality results in high throughput sequencing (HTS) techniques and other molecular techniques, extraction of high quality and pure DNA is essential [7,8].
Barnyard grass (Echinochloa colona) is an important weed in agriculture and is considered as one of the major troublesome weeds worldwide [9]. Among the various Echinochloa spp., E. colona is the most harmful and competing weeds for nutrients in field crops [10]. E. colona is an annual or perennial grass which is distributed throughout the warm regions of the world. It is common in areas of heavy rainfall and variable temperature. This is weed is highly competitive and difficult to manage in agricultural fields. Mechanical and cultural practices, as well as numerous herbicides, are used for its management around the world. The intensive and excessive use of glyphosate has resulted in the appearance of glyphosate-resistant E. colona biotypes [11]. The implementation of genomic tools could reduce the intensive use of herbicides for greater long-term sustainability in weed management The alkaloids, steroids, carbohydrates, glycosides, tannins, phenols and flavonoids contained in E. colona affect the quality of extracted DNA prerequisite for further molecular studies [6,12]. Moreover, the anionic contaminants present inhibit restriction enzymes and affect enzymatic analysis of the DNA [13].
High-quality DNA extraction from plant leaves is laborious, time-consuming, and costly. The use of commercially available kits still involves a high cost whilst providing a low yield of DNA [14]. Cetyltrimethyl ammonium bromide (CTAB) methods used for DNA extraction are modified depending upon the intracellular components, composition of cell walls and the histones [15]. Most of the weeds have dormant seeds, making them difficult to germinate, which may require variations in temperature and other conditions. For molecular studies such as PCR amplifications, an efficient and good quality DNA extraction method is required for next generation sequencing (NGS) and library preparations. Barnyard seeds are small and take a lot of time to germinate and grow uniformly. The goal of this study was to compare the relative efficacy of DNA extraction in barnyard plant leaf samples and the dry seeds using modified CTAB and a commercially available kit. High-quality DNA extraction is a prerequisite to weed genomics research. The present study was carried out to compare the quality and quantity of DNA isolated using two different tissue samples of barnyard under a modified CTAB and a commercially available kit.
Sample and material
Echinochloa colona weed samples were collected from infested areas of the states of Queensland and New South Wales, Australia. These samples were purified and stored in the at the Queensland Alliance for Agriculture and Food Innovation (QAAFI), The University of Queensland, Gatton Campus. For DNA extraction well-matured seeds from each accession were selected and raised in pots in controlled conditions maintained at 30/20°C (day/night temperature). The plants were raised to 3-4 leaf stage for extraction of DNA. The seed samples of Echinochloa colona were stored in zip-lock bags at 4°C. The DNeasy Plant Mini Kit was purchased from Qiagen, Valencia, CA, USA. The DNA ladder and PCR reagents were purchased from Promega, Ltd., Madison, Wisconsin, USA. Primers were synthesized from Integrated DNA Technologies (IDT), Ltd., Coralville, Iowa, USA.
Solution and reagents used
Extraction buffer was 2.5% cetyl trimethyl ammonium bromide (CTAB), 100 mM Tris HCl (pH 8.0), 1.5 M NaCl, 25 mM ethylenediamine tetraaceticacid (EDTA), 3% polyvinylpolypyrrolidone (PVPP). Isopropanol 70% ethanol (v/v); Phenol: Chloroform: Isoamyl alcohol (25: 24: 1, v/v); Chloroform: Isoamyl alcohol (24:1, v/v); 0.3 M sodium acetate (pH 5.4); TE (Tris/EDTA) buffer: 10 mM Tris-HCl (pH 8.0); and 1 mM EDTA (pH 7.4). Commercially available kit (DNeasy Plant Mini Kit, Qiagen, Valencia, CA, USA). Taq DNA polymerase, Taq DNA polymerase buffer, RNase, Nucleotides dNTPs (G, A, T, C), SSR primers, 20 mL 0.5 M EDTA, Agarose gel and Ethidium bromide.
Equipment
The equipment utilized was a grinder or pestle mortar; multichannel pipettes (Eppendorf AG, Hamburg, Germany); plate mixer; plate centrifuge; water bath or oven or heating block, any type that can maintain the temperature at 65°C; microcentrifuge; benchtop centrifuges with rotors if greater quantity of DNA is required; NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA); polymerase chain reaction (PCR); electrophoresis systems; and Gel Documentation System (Gel DocTM EZ BIO-RAD Laboratories, Inc., Hercules, California, USA).
DNA extraction of E. colona leaves and seeds using Qiagen kit method
Echinochloa colona DNA from plant leaves and dry seed samples was extracted using the DNeasy Plant Mini Kit. The plants were raised to 3-4 leaf stage for extraction of DNA. Harvested leaves were placed in glassine bags and stored in ice. About 100 mg of leaves were ground to a fine powder using a pre-chilled pestle and mortar after adding liquid nitrogen to make the leaves brittle as well as to stop DNAase and other enzyme activities. For DNA extraction from dry seed, 20 mg of seeds were ground to a fine powder in pestle and mortar in liquid nitrogen. Further, the manufacturer’s instructions were followed for DNA from both the samples of leaves and dry seeds.
NA extraction of barnyard leaves and seeds using CTAB method
The plants were raised to 3-4 leaf stage for extraction of DNA. Harvested leaves were placed in glassine bags and stored in ice. About 100 mg of leaves were ground to a fine powder using pre-chilled pestle and mortar after adding liquid nitrogen to make leaves brittle as well as to stop DNAase and other enzyme activities. Similarly, 20 mg of dry seeds were grounded to a fine powder in pestle and mortar in liquid nitrogen for DNA extraction from dry seeds. The powdered material of leaves, as well as seeds, were transferred to 1 ml of pre-warmed extraction buffer (CTAB buffer) and incubated for 60 minutes with occasional stirring. An equal volume of chloroform: isoamyl alcohol (24:1) was added in both the samples and slowly mixed by inverting the tubes for 10 minutes till it made a dark green emulsion. Both the samples were centrifuged for 10 minutes at 8000 rpm. The supernatant of both leaves, as well as seeds, were then transferred to another tube and again treated with chloroform: isoamyl alcohol (24:1), mixed slowly for 10 minutes and centrifugation was done. To the supernatant, 0.6 volume (400 µl) of ice-cold isopropanol was added and stored at 4°C for 1-2 hours. Centrifugation was done at 13000 rpm for 10 minutes at 4°C. The supernatant was discarded, and the pellets were washed with 70% ethanol (200 µl – 300 µl) to remove contamination and were then air-dried. DNA was dissolved in 300 µl TE Buffer and stored at 40 °C. For purification of DNA, 300 µl of RNase (10 mg/ml) was added to the samples and incubated for 1 hour at 37 °C in a water bath. An equal volume of phenol:chloroform:isoamyl alcohol (PCI) (25:24:1) was added in both the samples, tilted for 10 minutes and centrifuged at 13,000 rpm for 10 minutes. The supernatant was collected in another tube, 0.6 volume of chilled isopropanol was added, and centrifugation was done to get a pellet. DNA pellets of both samples were washed with 70% ethanol, air dried, and dissolved in TE (Tris-Cl, EDTA) buffer.
Quantitative and qualitative analysis of DNA
The quality of the isolated DNA was checked on 1% agarose gel. The concentration and relative purity of the isolated DNA were checked using Nanodrop ND-1000 (Agricultural Genomic Laboratory). DNA samples were diluted with distilled water and adjusted to 30 ng μL-1 [16]. The DNA extracted samples from leaves and seeds were stored at -20°C until further use.
PCR amplification
PCR amplification was performed in a 25 μL reaction mixture that contained approximately 2 μL of 30 ng genomic DNA, 0.5 μL (50 pmol) of each primer (Integrated DNA technologies, Coralville, Iowa, USA), 12.5 μL PCR master mix (Promega, USA), 400 μM of dNTP mix, 0.5 μL of 2.5 U/μL Taq DNA polymerase (Promega, USA), 2.5 μL of 3.0 mM MgCl2 and nuclease-free H2O. Amplification was performed with a thermal cycler (Eppendorf Germany). The primer of gene EPSPS encoding 5-enolpyruvoylshikimate-3-phosphate (EPSP) synthase, a key enzyme in the shikimic acid pathway [17,18]. An approximately 500 bp fragment of the EPSPS gene primers was amplified in an automated DNA thermal cycler with the cycle parameters as follows: 3 min denaturing at 94 °C; 40 cycles of 30 s denaturation at 94 °C, 30 s annealing at 56 °C and 1 min elongation at 68 °C, and a final extension for 7 min at 68 °C. PCR amplified products of all the primers were subjected to gel electrophoresis using 2.0% agarose gel in 1X TAE buffer at 100 V. The microsatellite markers were well resolved on 2% agarose gel with 3-hour run. The fragment sizes were determined by comparing with a 100 bp DNA ladder (Promega, USA) and the ethidium bromide stained gels were documented using Gel Documentation System (Gel DocTM EZ BIO-RAD, USA).
For all molecular studies, good quality DNA extraction and reliable measurement of concentration and purity are required [19]. In the present study, modified CTAB and DNeasy Plant Mini Kitmethods were used for isolation of high-quality DNA from E. colona leaves and dry seeds to optimize a rapid and inexpensive protocol. The main complications of plant DNA isolation were associated with the presence of polyphenolic compounds including polysaccharides. These compounds co-precipitate during the DNA isolation process and then affect the quality and quantity of extracted DNA [16]. In the present study total DNA isolated from E. colona plant leaves and dry seeds were checked by a NanoDrop ND-1000 Spectrophotometer. DNA quality was good and the ratio of absorbance at 260 nm to 280 nm was 1.8-2.1 and 1.7-2.2 in case of plant leaves using CTAB method and commercial kit, respectively. DNA quality was 1.8-2.2 and 1.7-2.3 in case of dry seeds with CTAB method and commercial kit, respectively which indicated insignificant levels of contaminating proteins and polysaccharides. In the present study total DNA isolated from leaves and dry seeds of E. colona using two different methods was analysed by agarose gel electrophoresis (Figure 1). DNA extraction using the modified CTAB method yielded high-quality DNA ranging from 1.7 to 2.3 showing that the DNA is free from polysaccharides, flavonoids, polyphenols and essential oils.

Figure 1. PCR amplification and gel electrophoresis
To test the suitability of the extracted DNA for use in molecular studies a PCR based analysis was carried out using microsatellite (SSR) markers (Table 1). Extracted high-quality DNA from E. colona leaves and dry seeds using CTAB as well as the commercial kit was found suitable for PCR amplification. Results indicated that the PCR amplifications were successful in all four samples with a primer EPSPS (Figure 2). The results also showed that the extracted DNA from both leaves and dry seeds were free from the interfering compounds indicating the potentiality of isolated DNA for further genomic studies of weeds. Although commercially available kits are effective in extraction of DNA free from compounds, a significant amount of DNA was lost from the samples [5,20]. The genomic DNA extraction using CTAB protocol for suppressing the phenolic compounds was also carried out in E. colona in other studies [16,21].
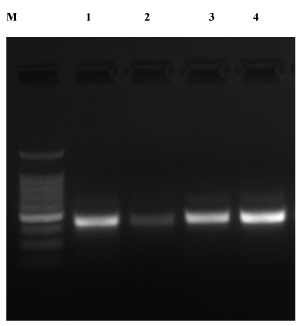
Figure 2. Agarose gel electrophoretogram of barnyard DNA amplified by PCR using EPSPS primers. Note: M: 100 bp DNA Ladder Marker. Lanes 1 and 2 PCR amplification of the DNA extraction from dry seeds using a modified CTAB method and commercial kit. Lane 3 and 4 PCR amplification of DNA extracted from plant leaves using modified CTAB and commercial kit
Table 1. PCR primers and their amplification result
Marker |
Forward primers (5’-3’) |
Reverse primers (5’-3’) |
Expected product size (bp) |
EC 1 |
F: ATTACTGGTCAGACGGAAAC |
R: GCAGTTATCTCCGTGGGCAC |
102-125 |
EC 2 |
F: GGCTCCAAACAAGGCAATTC |
R: TTCAGGGAATTTAGTACAAG |
150-177 |
EC 3 |
F: GAAAGGAAATGGGTTGGCTG |
R: CTTCGCACCATGATCTTCTC |
76-85 |
EC 4 |
F: AGTAGAAGGCTGCAAGAAGG |
R: TCTCAGCCCACTTTGTATAG |
167-181 |
EC 5 |
F: CAGAGCCTTCAATCATGGTG |
R: TGCTTCAAGTTCTAGGAGAC |
89-99 |
EG 302 |
F: ATTCGAACACCCATCAACCAAC |
R: GAAACAGAAGGGAGGTGTGCTG |
133-293 |
EG 306 |
F: TAAAACAAAACGACCGGCGTAA |
R: TCAATCATTTCAGCCTTCGGAT |
146-167 |
EG 320 |
F: CAACTCATAAGACAATTCAAAGGGTTT |
R: GCATCATTTAAGCATCAAAATGACA |
136-153 |
EPSPS |
F: AACAGTGAGGAYGTYCACTACATGCT |
R: CGAACAGGAGGGCAMTCAGTGCCAAG |
500 |
The results showed that the DNA extracted from dry seeds using modified CTAB and the commercial kit sufficiently supported the PCR amplifications of SSR marker EPSPS with 500 bp (Figure 2). The high-quality extracted DNA of E. colona usingy these methods can even be used for greater than 500 bp. DNA extraction from dry seeds can save time, manpower as well as money, which has high practical values in genomic and other molecular studies. This will also help in quickly screening other germplasm and related plant species material [22]. Most weed plants have small seeds and germination is reduced under artificial conditions. DNA isolation from dry seeds will save money, manpower and time. Moreover, the seeds can be easily preserved for a long time without specific preserving conditions and DNA can be extracted at any time to carry out the molecular studies in weeds. We also anticipate that this modified CTAB method of DNA isolation from dry seeds will be adequate for extracting high molecular weight and quality DNA from other species containing large amounts of essential oils and secondary metabolites. This will solve the problems caused by these compounds and essential oils during DNA extraction. DNA extracted from seeds as well as plant leaves using the modified CTAB protocol is cost-competitive, high throughput, and workable in routine laboratories and do not require any sophisticated equipment. Moreover, the protocol of DNA extraction from dry seeds using either a modified CTAB or commercial kit is valuable for resource savings in three ways i) at least one month time required for growing of seeds is eliminated, ii) field/greenhouse space is not required, and iii) manpower resources are saved. This will also enhance the efficiency of the scientists working with weed genomics. Moreover, for molecular studies such as genetic diversity analysis, genetic purity study, allele mining for important traits, high throughput sequencing studies etc. DNA extracted from dry seeds and plant leaves are equally important and immensely helpful in weeds.
DNA extraction is the first and primary step for molecular studies of plant species. Advanced molecular techniques depend upon the quantity and quality of extracted DNA from plants. Therefore, DNA extraction is an important and critical step in genomics and high throughput techniques to improve results. A number of protocols are available for extraction of DNA from plant leaves, but they are time-consuming and sometimes seeds do not germinate. Scientists can save valuable germplasm of plants that fail to germinate, as well as save time and manpower if a protocol for DNA extraction from dry seeds is available. It is therefore necessary to establish a less time consuming and inexpensive DNA extraction protocol for routine molecular work. Overall, the methods described here for DNA extraction from dry seeds are efficient and timesaving. Our results show that high-quality DNA can be isolated from plant leaf tissues using modified CTAB and commercial kits, and that DNA extracted from dry seeds will save time and other valuable resources.
Conceived and designed the experiments: B.S.C., R.S. Performed the experiments: B.S.C., R.S. Analyzed the data: R.S., B.S.C. Contributed reagents/materials/equipments/analysis tools: B.S.C. Wrote the paper: B.S.C., R.S.
The authors are thankful to Queensland Alliance for Agriculture and Food Innovation (QAAFI), The University of Queensland, Gatton, Queensland 4343, Australia and the Australian Government for providing Endeavour Executive Fellowship.
No potential conflict of interest was reported by the authors.
The work was supported by the Australian Government Endeavour Executive Fellowship Programme, Queensland Alliance for Agriculture and Food Innovation (QAAFI), The University of Queensland, Gatton, Queensland 4343, Australia.
- Stephen C, Weller Ray A, Bressan Peter B, Goldsbrough Tyler B, Fredenburg Hasegawa PM (2001) The effect of genomics on weed management in the 21st century. Weed Sci 49: 282-289.
- Stewart NC, Patrick J, Tranel, David P, Anderson J, et al. (2009) Evolution of weediness and invasiveness: Charting the course for weed genomics. Weed Sci 57: 451-462.
- Chacon-Cortes D, Griffiths LR (2014) Methods for extracting genomic DNA from whole blood samples: current perspectives. J Bioreposit Sci Appl Med 2: 1-9.
- Moore E, Arnscheidt A, Kruger A, Strompl C, Mau M (2008) Section 1 update: simplified protocols for the preparation of genomic DNA from bacterial cultures molecular microbial ecology manual. Springer, Netherlands pp: 1905-1919.
- Ariefdjohan MW, Savaiano DA, Nakatsu CH (2010) Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr J 9: 23.
- Sarwat M, Negi M, Lakshmikumaran M, Tyagi A (2006) A standardized protocol for genomic DNA isolation from Terminalia arjuna for genetic diversity analysis. Electron J Biotechnol 9: 86-91.
- Dilhari A, Sampath A, Gunasekara C, Fernando N, Weerasekara D, et al. (2017) Evaluation of the impact of six different DNA extraction methods for the representation of the microbial community associated with human chronic wound infections using a gel-based DNA profiling method. AMB Expr 7: 179.
- Li Z, Parris S, Saski CA (2020) A simple plant high-molecular-weight DNA extraction method suitable for single-molecule technologies. Plant Meth 16: 38.
- Mooney HA, Hobbs RJ (2000) Invasive species in a changing world. Island Press, Washington, D.C., USA.
- Walker S, Widderick M, Storrie A, Osten V (2004) Preventing glyphosate resistance in weeds of the northern grain region. In the 14th Australian weeds conference -Weed management: balancing people, planet and profit, Wagga Wagga, New South Wales, Weed Society of New South Wales, Sydney, Australia, pp: 428-431.
- Duke SO, Powles S B (2008) Glyphosate-resistant weeds and crops. Pest Manage Sci 64: 317-318.
- Sumitra S, Parul, Nidhi S (2018) Medicinal Potential of Weed Echinochloa colona (l.) Link: A Review. Intl J of Pharmaco and Phytochem Res 10: 98-102.
- Braid M, Daniels L, Kitts C (2003) Removal of PCR inhibitors from soil DNA by chemical flocculation. J Microbiol Methods 52: 389-93.
- Kang H, Cho Y, Yoon U, Eun M (1998) A rapid DNA extraction method for RFLP and PCR analysis from single dry seed. Plant Mol Biol Report 16: 1-9.
- Muhammad I, Zhang T, Wang Y, Zhang C, Qing M, et al. (2013) Modifi‑ cation of CTAB protocol for maize. Res J Biotech 8: 41-50.
- Danquah EY, Hanley SJ, Brookes RC, Aldam C, Karp A (2002) Isolation and characterization of microsatellites in Echinochloa (L.) Beauv. spp. Mol Ecol Notes 2: 54-56.
- Herrmann KM (1995) The shikimate pathway: early steps in the biosynthesis of aromatic compounds. Plant Cell 7: 907-919.
- Wang W, Xia H, Yang X, Xu T, Jiang Si H, et al. (2014) A novel 5-enolpyruvoylshikimate-3-phosphate (EPSP) synthase transgene for glyphosate resistance stimulates growth and fecundity in weedy rice (Oryza sativa) without herbicide. New Phytologist 202: 679-688.
- Aboul-Maaty NA, Oraby HA (2019) Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull Natl Res Cent 43: 25.
- Oates A, Bowling FL, Boulton AJ, McBain AJ (2012) Molecular and culture-based assessment of the microbial diversity of diabetic chronic foot wounds and contralateral skin sites. J Clin Microbiol 50: 2263-2271.
- Pipan B, Zupancic M, Blatnik E, Dolnicar P, Meglic V (2018) Comparison of six genomic DNA extraction methods for molecular downstream applications of apple tree (Malus x domestica). Cogent Food and Agri 4: 1.
- Liang H, Deng Y, Wang C, Xu X (2016) A high-throughput DNA extraction method from rice seeds. Biotech and Biotechnol Equip 30: 32-35.