Abstract
Introduction: The Wharton’s Jelly tissue possesses an attractive source for the isolation of mesenchymal stem cells (MSCs), that could be used in large scale clinical trials and in tissue engineering and regenerative medicine. However, prolonged cell culture and expansion, could affect their characteristics by causing genomic and epigenetic alterations. A possible solution to this problem would be the cryopreservation of the entire Wharton’s Jelly (WJ) tissue. The use of dimethyl sulfoxide for such a complex tissue is insufficient for its proper storage. Vitrification might be a feasible method for storage of more complex tissues. In order to find the optimum method for the storage of the WJ tissue different cryopreservation methods, including vitrification and conventional cryopreservation were applied in this study.
Methods: Vitrification was performed with either the use of a commercial available solution (“CryostorTM”) or VS55 a solution that can be used in non-equilibrium approach. Conventional cryopreservation was performed using a solution that combines 10% vol/vol DMSO and Fetal Bovine Serum. The evaluation of the cryopreservation methods was accomplished by the isolation of MSCs, growth kinetics, immunophenotypic analysis, multilineage differentiation, scratch wound assay and reactive oxygen species (ROS) quantification.
Results: The results clearly showed that the vitrification successfully preserved the WJ tissue at -196oC, yielding viable cells well characterized by mesenchymal stem cell properties. On the other hand, conventional cryopreservation did not achieve the same cell isolation rate when compared to vitrification methods.
Conclusion: In conclusion, the vitrification is a promising method for WJ tissue banking, allowing the isolation of well-defined MSCs for future MSC-based regenerative therapies.
Key words
MSCs, vitrification, cryopreservation, regenerative medicine, tissue engineering, ROS, liquid nitrogen, Whartron’s Jelly tissue
Introduction
The human umbilical cord, the vital link between the fetus and the mother, is comprised of two arteries and one vein. The intermediate space between the arteries and the vein is called WJ tissue [1]. This gelatinous matrix is well known as a rich source of the MSCs [2]. MSCs are considered as a mesodermal cell population with self-renewal capacity and capable for multilineage mesodermal differentiation [3]. Except from the WJ tissue, the MSCs are identified in bone marrow (BM), umbilical cord blood (UCB), amniotic fluid (AF) and adipose tissue (AT) [4]. Over the past of 20 years, it has been shown that MSCs have immunomodulatory, anti-inflammatory properties and their existence in the adult tissues and organs seems to be supportive for the tissue specific cells [5]. Indeed, MSCs are capable of expressing in the site of inflammation IL-6, IL-10, nitric oxide (NO), idoleamine 2,3 dioxygenase (IDO) and prostaglandin E2 (PGE2) and may cause apoptosis to T cells through Fas/Fas ligand and TNF receptor signaling pathway. Due to these properties the MSCs can now be used routinely as a co-therapy in immune related disorders such as multiple sclerosis (MS), amyotrophical lateral sclerosis (ALS) and Crohn’s disease [6].
Moreover, MSCs are characterized by the absence of HLA class II antigens, exerting in this way minimum immunoreactivity when transplanted allogeneically [7]. These characteristics, make them an ideal cell population for the regenerative medicine and tissue engineering approaches. The success of these approaches relies on the use of large number of well-defined MSCs that can be used under Good Manufacturing Practices (GMPs) [8]. Until now, in several studies, it has been described the use of MSCs in wound healing, development of tissue engineered blood vessels and extracellular matrices [9].
Nevertheless, while MSCs exist in the most adult tissues of the human body, only WJ-MSCs seem to be easily obtained with no invasive procedures from the umbilical cord, thus providing a large number of immune privileged cells. It is estimated that, the WJ tissue contains approximately 4.6 × 106 cells/cm [3]. In addition, the WJ-MSCs have less epigenetic modifications and characterized by greater proliferation and mesodermal differentiation abilities when compared to MSCs derived from the aforementioned sources. In this way, the WJ tissue is an attractive source of MSCs, that could be used in large-scale clinical trials [6].
Due to the large number of MSCs that is needed for the approaches of regenerative medicine and tissue engineering, in most times prolonged in vitro cell culture is needed. As a consequence, to this, shortening of telomere length and alteration of their morphologically, and self-renewal capacity as well as immunomodulatory and differentiation properties may occur.
The cryopreservation of MSCs, a widely applied method, could be used as an alternative approach in order to preserve the cells intact for a long-time period at -196oC [10]. Ideally, the MSCs could be recovered at any chosen time point and can be used in translation medicine [10]. Although, when these cells are recovered, it is necessary to be expanded in vitro, thus prolonging their culture even more. For this purpose, the successful cryopreservation of the WJ tissue and isolation of WJ-MSCs under desirable time points could be a hopeful strategy, avoiding the initial culture and being more beneficial to the translational medicine. Cryopreservation of cells is a well-established method for their long-term preservation [11]. On the contrary, the preservation of whole tissues at low temperatures by applying the cryobiological principles is still demanding considerable attention. Τhe dimethylsulfoxide (DMSO), which is used in the most cryoprotective solutions, cannot penetrate and protect efficiently the extracellular matrix (ECM) of the tissue, thus not reaching efficiently the resident cells [11]. As a result, the irreversible cell damage in combination with multiple morphological and structural changes in the tissue occurred due to the extracellular ice formation. WJ tissue is a rich source of glycosaminoglycans such as hyaluronic acid and chondroitin sulfate, where the MSCs rely on, indicating the use of a combination of cryoprotective agents that can protect both the extracellular matrix and cells. Under this scope, the vitrification seems to be a more feasible method for tissue cryopreservation, combining cryoprotective agents with high and low molecular weight. Furthermore, this method can be successfully applied when the cryoprotective agents in the system rapidly cooled, thus resulting in molecular motion “stasis” and the solidification of the system [12]. In addition, the devitrification temperature is lower when compared to the most conventional cryopreservation methods, providing greater recovery of viable cells. Additionally, the vitrification method is devoid of any animal or human sera, thus the vitrified WJ-MSCs could be broadly used in regenerative medicine and in large scale clinical trials. Until now, vitrification protocols have been applied successfully in long term storage of small model systems including human oocytes and embryos. This technology may further be used at scale-up experiments such as adipose tissue [13], blood vessels [14] and even more WJ tissue [15].
Nowadays, several studies [16] have provided evidence for the efficient cryopreservation and storage of the WJ tissue, though the most available protocols need further optimization in order to obtain high recovery rate of viable cells.
The aim of this study was the evaluation of two different cryopreservation protocols, vitirification and conventional cryopreservation for the proper storage of WJ tissue. For the vitrification protocol, two different solutions were tested, one commercially available solution (“CryostorTM”) and a second one that can be used in non-equilibrium vitrification approach (VS55). Accordingly, the conventional cryopreservation method relies on the use of a solution (which will be refereed as solution C), which contains 10% DMSO and fetal bovine serum (FBS). The efficiency of the different cryopreservation methods was assessed by isolation of MSCs from the preserved WJ tissue, cell viability, surface markers, multilineage differentiation, scratch wood assay and reactive oxygen species quantification in obtained MSCs from the cryopreserved WJ tissues.
Materials and methods
Collection of human umbilical cords
Fresh human umbilical cords (l=5-10 cm) were collected from normal deliveries (gestational ages 36-40 weeks) after informed consent form the mothers by experienced midwives trained in cord blood collection. The umbilical cords (n=10) were stored into Earl’s balanced salt solution (EBSS, Gibco, Life Technologies, Grand Island, USA) supplemented with 10 U/ml penicillin and 10 μg/ml streptomycin (Gibco, Life Technologies, Grand Island USA) at 4οC and processed within 24 hours from reception at the Hellenic Cord Blood Bank (HCBB). The collections were performed in accordance with the ethical standards of the Greek National Ethical Committee and were approved by our institution’s ethical board.
Wharton’s Jelly tissue isolation and cryopreservation protocol
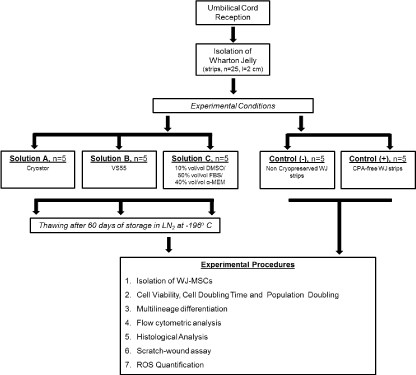
Initially a segment of the umbilical cord was cut using sterile surgical instruments, followed by briefly rinses in phosphate buffer saline 1x (PBS 1x) in order to remove excessive blood and blood clots. Then, the umbilical vein and arteries were removed and the WJ tissue was cut into strips (l=2 cm). A number of 5 samples of WJ tissue were placed into cryotubes and cryopreserved in each solution. The cryopreservation solutions used in this study, except the Solution A, consisted of the following compositions with α-Minimum Essentials Medium (α-MEM) as diluent.
Solution A: “CryostorTM” solution (BioLife Solutions, Monte Ville Parkway, USA)
Solution B: Vitrification solution (VS55), 3.10 M DMSO/3.10 M formamide/2.21 M 1,2-propanediol (Sigma Aldrich, St Louis USA) in Euro-Collins solution [17].
Solution C: 10% vol/vol DMSO (Sigma Aldrich, St Louis USA)/ 50% vol/vol FBS (Sigma Aldrich, St Louis USA).
The cryotubes containing WJ-strips were filled with precooled Solution A or B, rapidly cooled (43o C/min) to -100oC and then slowly cooled (3oC/min) to -135oC. Finally, the samples were stored in liquid nitrogen at -196oC. The cryotubes with WJ-strips, preserved with Solution C were placed in Mr Frosty containers, frozen overnight at a slow cooling rate of 1o C/min from room temperature to -80o C. Then transferred to cryoboxes, placed into liquid nitrogen (LN2) tanks at -196o C and stored for a time period of 60 days. Non-cryopreserved WJ-strips and WJ-strips stored in LN2 free of any cryoprotective agent (CPA-free), were used as negative and positive control groups respectively. A schematic diagram of the experimental procedure is presented in Figure 1.
2021 Copyright OAT. All rights reserv
Figure 1. Schematic representation of the experimental design.
Wharton’s Jelly thawing protocol
After 60 days of storage in LN2, the WJ-strips were thawed in order to isolate the WJ-MSCs. The cryotubes contained the WJ-strips were removed from the LN2 and immediately immersed in waterbath at 37oC, until the cryopreserved WJ-strips thawed properly. Finally, the WJ-strips were transferred to 50 ml polypropylene falcon tubes with 20 ml of PBS 1x and centrifuged at 500g for 6 min. The supernatant was discarded, and each WJ-strip was transferred to a 100 mm2 Petri dish until further processing.
Isolation of Wharton’s Jelly mesenchymal stem cells
Each WJ-strip was cut into small pieces (1-3 mm3) with the use of sterile surgical instruments and placed into 6-well plate (Costar, Corning Life, Canton, MA). Then, 1 ml of culture medium was added in each well and finally the 6-well plates were transferred in a humidified atmosphere with 5% CO2 at 37oC. After 15 days, microscopically examination was performed and upon reaching the sufficient number of adherent cells in 6-well plates, the cells were detached using 0.25% trypsin EDTA solution (Gibco), washed with PBS 1x, and re-plated into 75 cm2 flasks (Costar). On reaching 70-80% of confluency, the WJ-MSCs were trypsinized, washed and resuspended into the 175 cm2 flasks. The same procedure was repeated until the cells reached passage 5 (P5) of culture. The culture medium used for isolation and expansion of WJ-MSCs was α-Minimum Essentials Medium (α-ΜΕΜ,Gibco) supplemented with 15% fetal bovine serum (FBS, Gibco), 10 U/ml penicillin (Gibco),10 μg/ml streptomycin (Gibco) and 2mM L-glutamine (Gibco). The culture medium was changed twice every week and the cultures were maintained in a humidified atmosphere with 5% CO2 at 37o C.
Cell viability, doubling time and generation time of WJ-MSCs
WJ-MSCs were isolated from 5 independent WJ-strips of each experimental condition, continuously cultured from P1 to P5 and counted once they reached 80% confluency at each passage. Initially, 2 × 105 cells in 75 cm2 flask at P1 were plated. The determination of cell viability of WJ-MSCs was performed with the use of Trypan blue. Initially, the population doubling (PD) rate was determined at each passage using the following equation:
PD=log_10??(N/N0)?/log_10??(2)?
Additionally, the cell doubling time (CDT) from P2 to P8 was calculated by the classical formula:
CDT= log_10??(N/N0)?/log_10??(2)? x (T)
Where N is the number of cells at the end of the culture, N0 is the number of cells seeded, and T is the culture duration in hours.
In vitro multilineage differentiation
The in vitro multilineage differentiation capacity of WJ-MSCs of each experimental condition was assessed by adipogenic, osteogenic and chondrogenic differentiation. For this purpose, 12 × 104 cells/ well were seeded in 6-well plates (Costar) with culture medium. The next day, the culture medium was removed, followed by a brief wash with PBS 1x and adipogenic, osteogenic and chondrogenic induction was performed. The adipogenic differentiation of WJ-MSCs was committed with the use of basal medium (Mesencult, StemCell Technologies) supplemented with 10% of adipogenic stimulatory supplements (StemCell Technologies) for 25 days and assessed by staining of lipid vacuoles with Oil Red-O (Sigma-Aldrich) staining. WJ-MSCs were differentiated into osteogenic lineage using basal medium (Mesencult, StemCell Technologies, Vancouver, BC Canada) supplemented with 15% osteogenic stimulatory supplements (StemCell Technologies), 0.01 mM dexamethasone (StemCell Technologies) and 50 μg/ml ascorbic acid (StemCell Technologies). Osteogenic differentiation was assessed after 25 days with Alizarin Red S (Sigma-Aldrich) staining. Finally, chondrogenic differentiation was induced in a spheroid cultures using high glucose D-MEM supplemented with 0.01 mM dexamethasone (StemCell Technologies), 35 μg/ml ascorbic acid-2-phosphate (StemCell Technologies), 10 ng/ml transforming growth factor-β1 (Sigma-Aldrich), liquid medium supplement (ITS+ premix, Sigma-Aldrich) for 30 days. The pellets were fixed with 10% formalin (Sigma-Aldrich), paraffin embedded and cut into 5 μm sections. Chondrogenic differentiation was assessed with Alcian blue (Fluka, Sigma-Aldrich) staining.
Immunophenotypic analysis by flow cytometer
The immunophenotypic analysis of WJ-MSCs was performed at P2. The WJ-MSCs samples (n=3) of each experimental condition were analyzed for cell surface antigens by flow cytometry. Cells were tested for the MSCs markers CD90 (Thy-1), CD105 (endoglin), CD73 (ecto-5'-nucleotidase) [18]. Additionally, the WJ-MSCs were tested for CD29 (integrin subunit), CD19 (pan-B-cell marker), CD31 (pan-endothelial marker), CD45 (pan-hematopoietic marker), CD14 (monocyte marker), HLA-DR (HLA class II antigen), HLA-ABC (HLA class I antigen), CD14 (TLR-4 co-receptor), CD3 (T-cell co-receptor). More specifically, the cells were labeled with fluorescein isothiocyanate-conjugated anti-CD90, HLA-ABC, CD29, CD19, CD31, CD45. Epitopes CD105, CD73, CD44, CD3 and CD14, HLA-DR (all from Immunotech, Beckman Coulter, Marseille, France) were assessed with phycoerythrin-conjugated and PC5-conjugated mouse anti-human monoclonal antibodies respectively. The WJ-MSCs phenotypes were analyzed in Cytomics FC 500 (Beckman Coulter, Marseille, France) flow cytometer with the CXP Analysis software (Beckman Coulter).
Histological analysis
Each WJ-strip from the five experimental conditions was submitted to histological analysis. The WJ-strips (n=3) were fixed in 10% vol/vol neutral formalin (Sigma-Aldrich). Then, the fixed samples were embedded in paraffin and 5 μm cross sections were obtained. Finally, Hematoxylin and Eosin (H&E, Sigma-Aldrich) staining was performed. Images were obtained using Leica DM LS2 microscope with IC Capture v 2.2 software and processed with NIH Image J v1.50software.
Scratch-wound assay
WJ-MSCs of each experimental condition in a number of 12 × 104 cells/ well were plated in 6-well plates. The following day the scratch-wound assay was performed with a 20 μl pipette tip. Cell migration was captured after 24 and 48 h from the initial scratching using an inverted Leica DMIL with NIH Image J v1.50 software.
Reactive oxygen species quantification
The WJ-strips of the five experimental conditions were quantified for total antioxidants according to manufacturer’s instructions (Sigma-Aldrich). The total antioxidant’s concentration is relative to the levels of the ROS of cells. Briefly, the WJ-strips were incubated overnight with proteinase K 25 μg/ml (Sigma-Aldrich) at 55oC, following by inactivation of proteinase K at 95oC for 5 minutes. Then 100 μl of tissue lysates was loaded in each well of the 96 well plate (Costar). Another 100 μl of Cu2+ working solution was added to each well and incubation was performed for 90 minutes in dark place at room temperature. Finally, the absorbance of each sample was measured spectrophotometrically at 570 nm. The antioxidant’s content was determined by interpolation from a standard curve developed by Trolox Standard Solution. Additional controls used in ROS quantification, were freshly obtained WJ-MSCs (negative control group) and WJ-MSCs treated with 10% vol/vol DMSO (positive control group).
Statistical analysis
Statistical analysis was performed by using Graph Pad Prism v 6.01. Comparison in total cell number, cell viability, population doubling, cell doubling time and flow cytometric analysis of each experimental condition were performed with the unpaired nonparametric Mann-Whitney U-Test. Statistical significant difference between group values was considered when p-value was less than 0.05. Indicated values are mean ± standard deviation.
Results
Isolation and expansion of WJ-MSCs
WJ-MSCs were successfully isolated from all vitrified WJ-strips. Specifically, the WJ-MSCs were consistently isolated from the five of five WJ-strips vitrified with solution A (“CryostorTM”) and solution B (VS55). However, only in three of five WJ-strips which were cryopreserved with solution C (10% vol/vol DMSO/ 50% vov/vol FBS), the WJ-MSCs were isolated. On the other hand, no cells were able to be isolated from the CPA-free WJ-strips (positive control group).
The WJ-MSCs from vitrified WJ-strips (with solution A and solution B) exhibited spindle-shape morphology, when compared to cells isolated from non-cryopreserved WJ-strips (negative control group). The first plastic adherent cells appeared 7 days after culture at 37o C in humidified atmosphere and the 6-well plates were observed confluent after 15 days of the initial isolation (Figure 2). Moreover, these cells were able to be expanded and reached passage 5, in the same way as the cells that isolated from the non-cryopreserved WJ-strips (Supplementary File 1). On the contrary, the WJ-MSCs isolated from WJ-strips which were cryostored with solution C, resulted in a few viable cells, with low proliferation activity, no distinctive morphologic characteristics and in the end could not be expanded from the initial culture. Thus, only the MSCs obtained from vitrified WJ-strips with solution A and solution B were used for the next set of the experiments for this study.

Figure 2. Isolation of WJ-MSCs from stored WJ-strips at -196oC for a time period of 60 days. MSCs isolation from non-cryopreserved WJ-strips (negative control group) after 7 (A), 10 (F) and 15 (K) days of incubation at 37oC and 5% CO2. MSCs isolation from vitrified WJ-strips with solution A (Cryostor) after 7 (B), 10 (G) and 15 (L) days of incubation. MSCs isolation from vitrified WJ-strips with solution B (VS55) after 7 (C), 10 (H) and 15 (M) days of incubation. MSCs isolation from cryopreserved WJ-strips after 7 (D), 10 (I) and 15 (N) days of incubation, where only few viable cells were appeared. MSCs isolation of stores WJ-strips at -196o C without any CPA (positive control group) after 7 (E), 10 (J) and 15 (O) days, where no cells appeared. Original magnification 10x, scale bars 100 μ
In order to fully determine the characteristics of the obtained WJ-MSCs, we proceeded in the estimation of cell viability, population doubling and cell doubling time. Notably, by the end of P5, the total number of cells obtained from vitrified WJ-strips, surpassed 2 x 107 cells (Figure 3A). The mean viability of MSCs (P2 to P5) derived from cryostored WJ-strips with solution A and B ranged between 87.4 ± 3.9% to 89.4 ± 3.6% and 89.4 ± 1.9% to 91.4 ± 2.2% respectively (Figure 3B). Compared to non-cryopreserved WJ-strips, the mean cell viability was slightly decreased in the cryostored WJ-strips with solution A and B. These data indicated a statistical significance difference (p<0.01) in the mean cell viability between cryostored and non cryostored WJ-strips (Figure 3B). In the same way, the average PD and CDT from P2 –P5 for WJ-MSCs derived from WJ-strips with solution A ranged between to 3.8 ± 0.2 to 9.0 ±2.1 and 83.2 ± 4.6 to 196.1 ± 47.5 h respectively (Figure 3C and 3D). Additionally, for the vitrified WJ-strips the average PD and CDT ranged between to 3.8 ± 0.13 to 9.1. ± 2.0 and 85.6 ±2.7 h to 199.4 ± 45.4 h (Figure 4). No statistical difference was observed in the average PD and CDT between cryostored and non-cryopreserved WJ-strips.
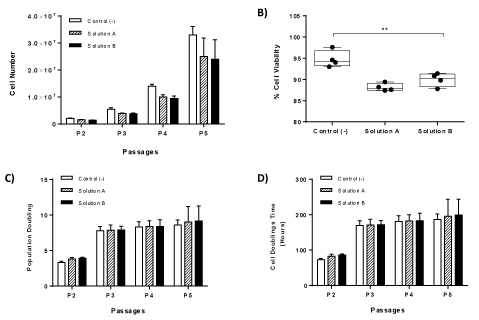
Figure 3. Kinetics characteristics of isolated MSCs. Total cell number of MSCs until reaching passage 5 (A). Measurement of % cell viability of WJ-MSCs until reaching passage 5 (B). Calculation of Population doublings of WJ-MSCs until reaching passage 5 (C). Calculation of Cell Doubling Time of WJ-MSCs until reaching passge 5 (D). Statistical significant difference was showed only in % cell viability between all study groups, p<0.01.
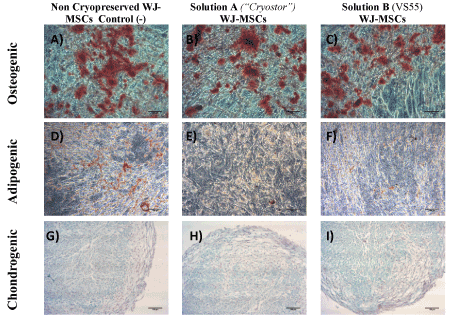
Figure 4. Multilineage differentiation of WJ-MSCs towards to osteogenic, adipogenic and chiondrogenic lineages stained with Alizarin Red S, Oil Red-O and Alcian blue stainings respectively. MSCs from non-cryopreserved WJ-strips (negative control group) successfully differentiated to osteogenic (A), adipogenic (D) and chondrogenic (G) lineages. MSCs from vitrified WJ-strips with solution A (“Cryostor”) successfully differentiated to osteogenic (B), adipogenic (E), and chondrogenic lineages (G). MSCs from vitrified WJ-strips with solution B (VS55) successfully differentiated to osteogenic (C), adipogenic (F), and chondrogenic lineages (I). Original magnification 10x, scale bars 100 μm.
Multilineage differentiation of MSCs
The differentiation ability of WJ-MSCs to mesodermal cell lines including osteogenic, adipogenic and chondrogenic lineages was assessed in cells that were obtained from the WJ-strips vitrified with Solution A and B and compared with the noncryopreserved WJ-strips. All samples differentiated successfully to osteogenic, adipogenic and chondrogenic lineages. Regarding the osteogenic induction, the differentiated MSCs were stained positively with the Alizarin Red S staining indicating the production of calcium deposits (Figure 4). In addition, the differentiated MSCs towards to adipogenic lineage were stained positively with Oil-red O staining. Finally, after 21 days of exposure to chondrogenic inducing factors, the MSCs from cryostored and non-cryostored WJ-strips exhibited glycosaminoglycan (GAG) production, that was visible with the Alcian blue staining (Figure 4). No significant differences were observed in the intensity of the above stains in all experimental groups that was tested.
Immunophenotypic analysis
The MSCs obtained from the vitrified WJ-strips with solution A and B expressed the same surface markers as the MSCs isolated from the non-cryopreserved WJ-strips. More specifically, flow cytometric analysis showed positive expression for β1 integrin subunit CD29 and matrix receptors CD90, CD105, CD73 and HLA- ABC and negative expression for CD19, CD3, CD31, HLA-DR and CD45. Representable diagrams of the flow cytometric analysis are shown in Figure 5. No statistical significant difference in the expression levels of the above surface antigens was observed between all MSCs samples (Supplementary File 2).
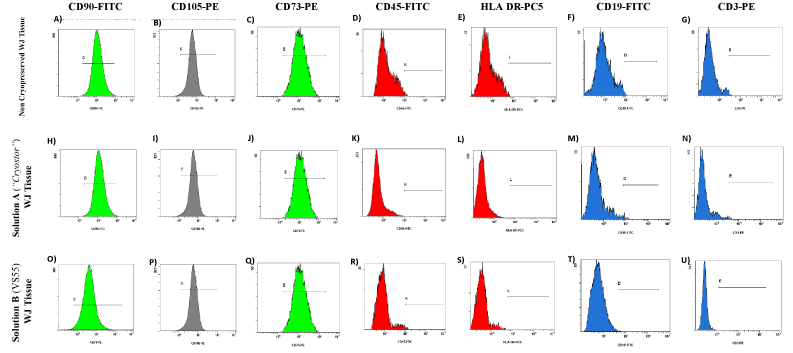
Figure 5. Flow cytometric analysis of WJ-MSCs derived either from non-cryopreserved or vitrified WJ-strips with solution A (“Cryostor) and B (VS55). WJ-MSCs derived from non-cryopreserved and vitrified WJ-strips with solution A and solution B showed strong expression of CD90 (A,H,O), CD105 (B,I,P), CD73 (C,J,Q) and weak expression of CD45 (D, K, R), HLA DR (E,L,S), CD19 (F,M,T) and CD3 (G,N,U).
Histological analysis
Histological analysis of WJ-strips was performed for further evaluation of the storage methods that were applied. More specifically the vitrified WJ-strips with solution A and B presented a dense gelatinous extracellular matrix, with no observable destruction and damage of the resident cells. Morphologically the WJ-strips ECM did not present any significant difference from the non-cryopreserved WJ-strips. On the contrary, cryopreserved WJ-strips and WJ-strips that were stored without any CPA at -196oC characterised by large intermediate spaces in the ECM indicating its extensive damage and only by few MSCs. These spaces are quite characteristic in stored tissues at low temperatures and occurred due to the extracellular ice formation during the cryopreservation procedure.
Scratch-wound assay
The scratch-wound assay was used to evaluate the migration ability of the MSCs. Specifically, the MSCs obtained either from the vitrified WJ-strips presented the same migration activity as the non-cryopreserved MSCs. Finally, after 48h complete closure of the initial occurred gap was observed in all samples indicating their successful cell adhesion and migration.
ROS quantification
Non-cryopreserved WJ-strips and vitrified WJ-strips (with solution A or solution B) presented lower levels of antioxidants when compared with the CPA-free WJ-strips. However, the WJ-strips cryopreserved with solution C were characterized by the same levels of total antioxidants as the CPA-free samples and by higher levels of ROS when compared with the vitrified WJ-strips with solution A and B. Furthermore, the antioxidant levels of cryopreserved WJ-strips with solution A and B were statistical significant lower when compared with MSCs treated with 10% DMSO (positive control group). Additionally, WJ-strips cryostored with solution C and CPA-free samples presented the same antioxidant levels as the positive control group and higher when compared with MSCs isolated from non-cryopreserved WJ-strips which was statistical significant (Figures 6- Figure 8 and Supplementary File 3).
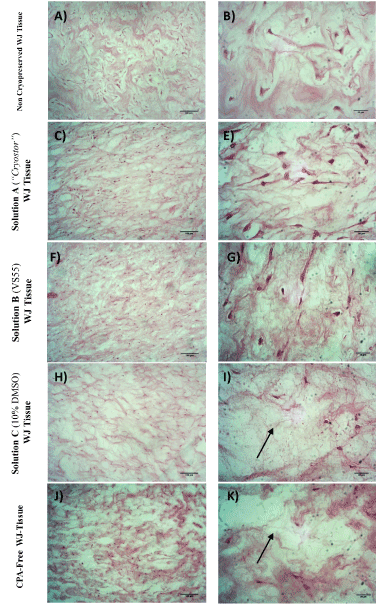
Figure 6. Histological analysis of WJ-strips after the application of vitrification or cryopreservation protocols stained with H&E. Fresh, non-cryopreserved WJ tissue characterized by dense gelatinous ECM with a great number of cell (A,B). Vitrified WJ-strips with solution A (C,E) and solution B (F,G) presented well retained structure as the non cryopreserved WJ tissue. WJ tissue cryopreserved with solution C (H,I) or stored without any CPA at -196oC (J,K) characterized by extensive destruction as indicated by the arrows. Images A,C,F,H and J acquired by original magnification 10x, scale bars 100x. Images B,E,G,I and K acquired by original magnification 40x and scale bars 50 μm.
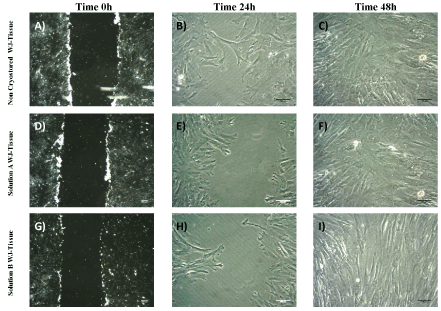
Figure 7. Scratch wood assay performed by WJ –MSCs. Total closure of the initial gap was accomplished after 48 h of incubation at 37oC and 5% CO2. WJ-MSCs derived either from non-cryopreserved (A,B,C) or vitrified WJ-strips with solution A (D,E,F) and B (G,H,I) migrated and filled the occurred gap at the same time point. Images A,D and G acquired by original magnification 2.5 x and scale bars 200 μm. Images B,C, E,F,H and I acquired by original magnification 10x and scale bars 100 μm.
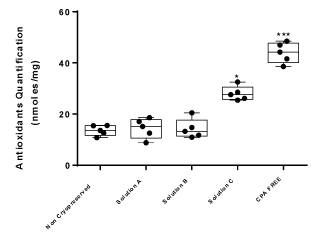
Figure 8. Antioxidant quantification of WJ-strips. Five different experimental conditions were tested for the total antioxidant amount in WJ-strips. The WJ-strips vitrified with solution A and B or cryopreserved with solution C were compared with non-cryopreserved or stored WJ-strips without any CPA. Statistical significant difference was occurred between WJ-strips with solution C and the rest of the samples, p<0.05. WJ-strips stored without any CPA also presented statistical significant difference from the rest of the samples, p<0.01.
Discussion
The aim of this study was to evaluate different cryopreservation methods that could be used for the long-term storage of the WJ tissue. Currently, the conventional cryopreservation methods are very difficult to be applied for the proper storage of WJ tissue at low temperatures [19]. Until now, several attempts with the use of different cryoprotective solutions have been performed in a wide variety of tissues in order to establish the cryopreservation and storage conditions [20]. Among them, the vitrification approach might be the best current cryopreservation method, combining cryoprotective agents with low and high molecular weight. As a result, the ECM could be preserved efficiently, while the tissue cells are recovered by higher viability rates when compared with the conventional cryopreservation methods.
In this study, three different cryopreservation solutions, a commercially available solution (“CryostorTM”), a vitrification solution (VS55) and a conventional cryopreservation solution with 10% vol/vol DMSO were used and evaluated for the successful storage of WJ-strips at -196oC for a time period of 60 days. After this time period, the WJ-strips were thawed quickly and proceeded to the isolation of MSCs. More specifically, the MSCs were obtained, expanded and passaged successfully from the vitrified WJ-strips either with solution A (“CryostorTM”) or solution B (VS55). Morphologically, these cells were spindle-shaped without any observable differences when compared to MSCs obtained from non-cryopreserved WJ-strips (negative control group). On the other hand, only few viable cells were isolated from the conventionally cryopreserved WJ-strips (with solution C), and these were not able to be further expanded and passaged. These results were in accordance with a previous study of Roy and his colleagues [21], where the same difficulties regarding the isolation of viable cells were observed. Under this scope, it can be assumed that DMSO cannot penetrate efficiently the extracellular matrix of the tissue, favoring the development of endogenous ice formation and possible damaging of the tissue resident cells. On the contrary, the vitrification protocols could potentially store properly the tissue at very low temperatures by combining low and high molecular weight cryoprotective agents. According to cryobiological principles, the low molecular weight CPAs can penetrate efficiently the ECM reaching the cells, contributing in this way to the proper preservation of cells. The high molecular weight CPAs are responsible for the preservation of the ECM, thus not allowing the ice crystal formation, reducing the tissue thermomechanical stress and further protecting the tissue resident cells from any damage caused by the low temperatures.
Next, we proceeded in the calculation of the mean cell number, CDT, PD and cell viability only in MSCs obtained from the vitrified samples (preserved with “CryostorTM” and VS55) in order to evaluate thoroughly the impact of the cryopreservation solutions to this tissue. The MSCs derived from the vitrified WJ-srtips, successfully reached passage 5 with an average number of 2.5 x 107 cells for solution A and 2.4 x 107 cells for solution B whereas the average number of cells obtained from non-cryopreserved WJ-strips reached 3.3 x 107 cells. Despite this difference in the average cell number between the different experimental conditions, no statistical significant difference was observed as it was calculated by Kruskal-Wallis test. Regarding the mean CDT and PD at passage 5 of vitrified WJ-strips was 196.1 ± 47.5 h, 9.0 ± 2.1 respectively for solution A and 199.4 ± 45.4 h, 9.1. ± 2.0 respectively for solution B. Based on the trypan blue assay, the mean cell viability of the above specimens was 89.4 ± 3.6% for MSCs derived from WJ-strips with solution A and 91.4 ± 2.2% for solution B.
The immunophenotyping analysis revealed that the MSCs were more than 80% positive for CD90, CD105, CD73, CD29, CD44 and less than 3% negative for CD3, CD31, CD14, CD45 and HLA-DR. No statistical significant difference was occurred in flow cytometric analysis results when compared to the MSCs obtained from non-cryopreserved samples.
Further evaluation of the different cryopreservation methods to the isolated MSCs regarding their multilineage differentiation capacity was performed by differentiating them towards to adipogenic, osteogenic and chondrogenic lineages with the use of defined growth factors. Indeed, the MSCs achieved to develop calcium deposits and lipid vacuoles under osteogenic and adipogenic induction conditions respectively as identified by Alizarin Red S and Oil Red O stainings. In addition, under chondrogenic induction conditions the MSCs succeeded to produce sulfated glycosaminoglycans, key macromolecules of the cartilage ECM, indicating the successful differentiation towards to the chondrogenic lineage. Currently, the previous results clearly showed the retaining of the mesenchymal stromal cell properties in cells that were obtained from vitrified tissues. Until date, a majority of studies involve the use of conventional cryopreservation methods for storage of WJ tissue at low temperatures with contradictory results. Despite this significant difference in the experimental procedure, our results regarding the immunophenotypic analysis and multilineage differentiation of MSCs seem to be in accordance with previously published studies [5]. In the study of Shivakumar and his colleagues [22], the successful isolation, expansion and multilineage differentiation of MSCs were quite similar to our results with the only difference that in our study the optimum cryopreservation protocol for storage of WJ tissue was the vitrification.
Moreover, despite our efforts, no MSCs were obtained from WJ tissue, when a conventional cryoprotective solution with 10% vol/vol DMSO was applied. Concerning this, and in order to completely find the exact reason of this issue, histological analysis of the WJ tissue with H&E staining was performed. The WJ tissue is a dense gelatinous matrix where the MSCs relies on [15]. As it was revealed by H&E staining, this formation retained after the application of the vitrification protocol either with solution A or solution B and without exhibiting any significant morphologically differences from the non-cryopreserved WJ tissue. On the other hand, the WJ-strips that were cryopreserved with solution C (conventional method) exhibited extensive destruction of the ECM with accompanied damage of the resident cells. This morphology is quite characteristic when extracellular ice formation is occurred during the cryopreservation, indicating the unsuccessful penetration of the cryoprotective agent to the tissue.
Scratch-wound assay results showed that MSCs derived from vitrified samples characterized by the same migration capacity as the negative control group. The initial occurred gap, closed after 48 hours, a time period that has been described by other groups in literature. The migration ability of the MSCs is very important especially when those cells intended to be used for wound healing and regenerative medicine approaches.
Until today, several groups worldwide have tried to cryopreserve and store properly the WJ tissue at low temperatures. However, in the study of Da-Croce and her colleagues [23] was shown that the slow-cooling vitrification protocol was responsible for the extensive cell damage, thus no cells were obtained from the vitrified samples. On the contrary to this group, our study provided clear results regarding the enhanced survivability of MSCs from vitrified samples when compared with cryopreserved samples. A better knowledge to this is provided by the ROS quantification experiments that have been performed by our group. Both vitrified WJ-strips characterized by the same content of antioxidants as MSCs obtained from fresh tissues and non-cryopreserved WJ-strips. Statistical significant difference in the antioxidant content was occurred between the vitrified and cryopreserved samples. Moreover, the antioxidant content of cryopreserved WJ-strips was slightly lower from the WJ-strips that were stored in liquid nitrogen without any cryoprotective agent. In this study, elevated ROS levels could be induced by the high thermomechanical stress that was occurred to the MSCs during the application of the cryopreservation procedure [19]. It is known that high ROS levels induce further mitochondrial damage, responsible for inducing cell apoptosis via cytochrome c release and caspase 3 activation. This might be the primary reason for the low amount of isolated cells from the cryopreserved WJ-strips. The huge discrepancy that has occurred between the two studies might reflect different experimental parameters that were applied during the vitrification protocol of the WJ-strips. Possibly, different time points set or slightly different combination of cryoprotective agents used in the vitrification protocol might harm irreversible the WJ tissue.
The WJ is a very promising source of MSCs that could be used in translational medicine either for autologous or heterologous applications. In the most cases of tissue cryopreservation, the cryosolutions consist of 10% vol/vol DMSO with FBS. The addition of FBS in cryosolutions is needed for the proper cryopreservation, although bovine proteins of FBS impedes the utility of the stored cells or tissues for clinical use. Additionally, the typical DMSO concentration, used for cryopreservation, range between 5-20% vol/vol and may not be efficient to preserve complex tissues, thus damaging even more the resident cells. On the other hand, the vitrification method has been used successfully for the preservation of oocytes in the past, and its applications could be extended even more such as in the cryopreservation of most complex tissue like the WJ tissue.
Conclusion
In conclusion, the proper preservation and storage of WJ tissue is of paramount importance and can be performed either with a commercially available solution or by application of non-equilibrium approach. The WJ tissue could be used as an alternative source of ready to use well-defined MSCs without loss of stem cell characteristics. Until now, the bone marrow is the most preferred source for the isolation of MSCs, which are used widely in in regenerative medicine and large scale clinical trials. However, since there is low percentage of MSCs in the whole bone marrow (0.01-0.001%) [4], in most cases prolonged cell expansion and culture is required. The vitrification method might be a feasible method for the proper storage of WJ tissue, and the isolation of MSCs can be performed at any desired time without the need of the prolonging cell culture.
Authorship and contributorship
Mallis Panagiotis (first author) carried out the experiments regarding the cryopreservation of Wharton Jelly tissue, isolation, expansion, quality control of MSCs and histological analysis. Additional, wrote the publication and performed the statistical analysis of the experiments. Georgiou Eleni contributed in the experiments regarding the cryopreservation of Wharton Jelly tissue, isolation and tri-lineage differentiation of MSCs Michalopoulos Efstathios designed and supervised the overall study. Catherine Stavropoulos Giokas supervised and approved the overall study.
References
- Joerger-Messerli MS, Marx C, Oppliger B, Mueller M, Surbek DV, et al. (2016) Mesenchymal Stem Cells from Wharton’s Jelly and Amniotic Fluid. Best Pract Res Clin Obstet Gynaecol 31: 30-34. [Crossref]
- Kim DW, Staples M, Shinozuka K, Pantcheva P, Kang SD, et al. (2013) Wharton’s Jelly-Derived Mesenchymal Stem Cells: Phenotypic Characterization and Optimizing Their Therapeutic Potential for Clinical Applications. Int J Mol Sci 14: 11692–1171. [Crossref]
- Fong CY, Richards M, Manasi N, Biswas A, Bongso A (2007) Comparative growth behaviour and characterization of stem cells from human Wharton’s jelly. Reprod Biomed Online 15: 708-718. [Crossref]
- Fellows CR, Matta C, Zakany R, Khan IM, Mobasheri A (2016) Adipose, Bone Marrow and Synovial Joint-Derived Mesenchymal Stem Cells for Cartilage Repair. Front Genet 7: 213. [Crossref]
- Bernardo ME, Cometa AM, Pagliara D, Vinti L, Rossi F, et al. (2011) Ex vivo expansion of mesenchymal stromal cells. Best Pract Res Clin Haematol 24: 73-81. [Crossref]
- Hanley PJ, Mei Z, da Graca Cabreira-Hansen M, Klis M, Li W, et al. (2014) Manufacturing mesenchymal stromal cells for phase I clinical trials Cytotherapy 15: 416-442. [Crossref]
- Fracchiolla NS, Fattizzo B, Cortelezzi A (2017) Mesenchymal Stem Cells in Myeloid Malignancies: A Focus on Immune Escaping and Therapeutic Implications. Stem Cells Int: 13.
- Izadpanah R, Kaushal D, Kriedt C, Tsien F, Patel B, et al. (2008) Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res 68: 4229-4238. [Crossref]
- Li B, Moshfegh C, Lin Z, Albuschies J, Vogel V (2013) Mesenchymal stem cells exploit extracellular matrix as mechanotransducer. Sci Rep 3: 2425. [Crossref]
- Gurruchaga H, del Burgo LS, Garate A, Delgado D, Sanchez P, et al. (2017) Cryopreservation of Human Mesenchymal Stem Cells in an Allogeneic Bioscaffold based on Platelet Rich Plasma and Synovial Fluid. Sci Rep: 7.
- Protocols F (2007) Cryopreservation and freeze-drying protocols.
- Hunt CJ (2011) Cryopreservation of Human Stem Cells for Clinical Application: A Review. Transfus Med Hemother 38: 107-123. [Crossref]
- BKocan B, Maziarz A, Tabarkiewicz J, Ochiya T, Banas-Zabczyk A (2017) Trophic Activity and Phenotype of Adipose Tissue-Derived Mesenchymal Stem Cells as a Background of Their Regenerative Potential. Stem Cells Int: 1653254. [Crossref]
- Jung Y, Ji H, Chen Z, Chan HF, Atchison L, et al. (2015) Scaffold-free, Human Mesenchymal Stem Cell-Based Tissue Engineered Blood Vessels. Sci Rep 5: 15116. [Crossref]
- Jadalannagari S, Converse G, McFall C, Buse E, Filla M, et al. (2017) Decellularized Wharton’s Jelly from human umbilical cord as a novel 3D scaffolding material for tissue engineering applications. PLoS One 12: e0172098. [Crossref]
- Baicu S, Taylor MJ, Chen Z, Rabin Y (2008) Cryopreservation of carotid artery segments via vitrification subject to marginal thermal conditions: Correlation of freezing visualization with functional recovery. Cryobiology 57: 1-8. [Crossref]
- Dahl SL, Chen Z, Solan AK, Brockbank KG, Niklason LE, et al. (2006) Feasibility of vitrification as a storage method for tissue-engineered blood vessels. Tissue Eng 12: 291-300. [Crossref]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, et al. (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8: 315-317. [Crossref]
- Wowk B (2010) Thermodynamic aspects of vitrification. Cryobiology 60: 11-22. [Crossref]
- Sheridan WS, Duffy GP, Murphy BP (2013) Optimum Parameters for Freeze-Drying Decellularized Arterial Scaffolds. Tissue Eng. Part C Methods 19: 981-990. [Crossref]
- Roy S, Arora S, Kumari P, Ta M, (2014) A simple and serum-free protocol for cryopreservation of human umbilical cord as source of Wharton’s jelly mesenchymal stem cells. Cryobiology 68: 467-472. [Crossref]
- Shivakumar SB, Bharti D, Subbarao RB, Jang SJ, Park JS, et al. (2016) DMSO- and Serum-Free Cryopreservation of Wharton's Jelly Tissue Isolated from Human Umbilical Cord. J Cell Biochem 117: 2397-2412. [Crossref]
- Da-Croce L, Gambarini-Paiva GH, Angelo PC, Bambirra EA, Cabral AC, et al. (2013) Comparison of vitrification and slow cooling for umbilical tissues. Cell Tissue Bank 14: 65-76. [Crossref]