Abstract
This investigation was aimed at assembling different genetic building blocks to produce a focused DNA sensor to detect proteins related to glucose in blood, in order to diagnose early diabetes using synthetic biology and conventional molecular biology. A glucose DNA sensor was constructed in Saccharomyces cerevisiae using genomic and synthesized sequences and were tested in both, In vivo using human blood plasma sampled, and In vitro using cultural media. The results were based on fluorescence intensity of the DNA sensor and compared with clinical methods. The DNA sensor was highly sensitive when mixed with plasma samples from different patients (i.e., diabetic, pre-diabetic, and normal) showing high fluorescence, and was able to detect a wide concentration range of glucose equivalent to clinical glycaemia values. Expression of proteins pertaining to glucose metabolism production was determined by 2D-DIGE gel electrophoresis-maldi analysis. The glucose sensor produced results less than a minute after being mixed with a drop of a human blood sample. Our results highlight the advantages of using constructed DNA sensors to detect glucose in blood for early diagnosis of diabetes.
The yeast DNA glucose sensor was assembled successfully with standardized genetic parts and was able to detect low levels equivalent to clinical glycaemia (<140 mg/dL), as well as a higher equivalent level of glycaemia (>200 mg/dL). Thus, our sensor can be used for early diagnosis of diabetes or pre-diabetic conditions, thereby allowing for earlier clinical intervention. The direct correlation between levels of glucose and the intensity of fluorescence of the DNA sensor shows the advantage of using this technology in order to identify specific types of diabetic patients. Sometimes, it is difficult to establish accurate correlations with different methods.
Key words
DNA glucose sensor, glucose, fluorescence, plasmid, glycaemia, diabetes, synthetic biology
Introduction
There have been large number of publication regarding spontaneous emissions of light from cells. This is based on ultra-weak photon emission (UPE), which is usually referred as biological luminescence or bio-photons. Usually, cells are capable to emit one to thousands of photon/s cm2 over a basal equipment inherent noise level. Cells are able to increase their UPE intensity level when exposed to physiological changes, including environment stress, such as presence of chemicals [1].
A biosensor is a device that detects, transmits, and records information regarding physiological or biochemical changes. It functions as a probe that integrates a biological component with an electric transducer, converting a biochemical signal into a quantifiable electrical response [2].
Therefore, a microbial DNA sensor can be classified as a biosensor. Sensors have a variety of applications in everyday life, including as components of machines and robotic cars, in medicine, in environmental care, in industry, in clinical diagnostics, and in various other areas. Many different types of sensors have been developed. Physical or electrical sensors, which detect physical properties such as temperature, pressure, humidity, and movement, are widely used; chemical sensors have been developed to detect different molecules, including gases such as oxygen and ammonia, or specific molecules in solution [3,4]. These produce a variety of measurable responses. For example, biological, cellular, or molecular sensors can detect organic compounds that may be useful as biomarkers [5,6]. Proteins such as glycoproteins and/or glucose transport proteins, as well as genes (i.e., GLUT) have been used as markers to indicate glucose presence in human blood [7]. These types of sensors are versatile analytical tools that offer advantages over classical analytical methods due to their inherent specificity, selectivity, and simplicity [5]. Most biological and cellular sensors use membrane and cellular proteins that recognize and take up biomarkers from a medium [6]. However, DNA has also been used in molecular biosensors [8,9].
Biomarkers for detecting and diagnosing diseases include physical symptoms, mutated DNA and RNA, secreted proteins, processes such as cell death or proliferation, and serum concentrations of small molecules such as cholesterol or, in the case of diabetes, glucose [6].
Currently, the most common methods to diagnose diabetes use enzymatic reactions to determine glucose levels [10,11]. These methods measure glucose in whole blood because erythrocytes contain high concentrations of glucose [12]. The methods include the use of glucose oxidase, hexokinase, and glucose dehydrogenase [13-15]. The products of these reactions with blood sugar can be determined using colorimetric and spectrophotometric assays or by measuring the electric current produced in the enzyme reactions; the latter is the approach used in most commercially-available glucose meters [16]. These methods are accurate and sensitive, and some are capable of detecting glucose concentrations ranging from 0 to 500 mg dL-1 for laboratory assays and from 20 to 500 mg dL-1 for home monitoring. However, current methods can provide false high values of glucose in the presence of interfering environmental conditions or in patients undergoing certain medical treatments [17,18]. Incompatibility of meters and strips may also lead to uncertainty, which is another persistent problem associated with current methods for measuring glucose levels.
The cost of current methods is also an impediment, where prices range from about $0.35 to $1 for each strip used in home monitoring sensors. Type 1 diabetics may test as often as 4 to 10 times a day, although type 2 diabetics typically test less frequently. In clinical laboratories, the cost of glucose quantification can range from $3 to $5, although in some countries, such as the United States, the cost can exceed $100. The cost of the analysis also depends on the type of analytical technique used. In the case of insulin analysis, prices range from $50 to $130 in the us and about $25 in some Latin American countries. Diabetes diagnostic tests that include the detection and quantification of both glucose and insulin can cost between $190 and $350, which is highly cost-prohibitive.
Other methods for detecting diabetic conditions based on biomarkers other than glucose are being developed. For example, the use of a powered laser can detect the presence of diabetes mellitus by monitoring physical changes caused by that disease in a patient’s eye [19]. In another example, immunology-based methods that detect the presence of glutamic acid decarboxylase (GAD) and thyroid peroxidase can be used to diagnose diabetes [20,21]. However, these techniques are costly and difficult to perform and evaluate at home, once the diabetic condition has already been established in the body.
In view of all the issues described above, a need exists for the development of a new, cost-effective, and accurate technology for detecting glucose and insulin levels in the early stages of diabetes in order to prevent or treat the disease. We applied the concepts of synthetic biology in combination with existing scientific methods to produce new a glucose detector. Effective use of constructed molecular sensors produced by synthetic biology to enhance the detection of metals has been reported [22]. The efficacy of in combining existing scientific methods with synthetic biology has been demonstrated [8]. Our sensor was constructed with various genetic parts using genomic and synthesized sequences. The yeast DNA sensor described here effectively detects glucose in human blood samples.
Materials and Methods
Construction of DNA Glucose Sensor
Synthesized DNA genetic parts from Clonetex Systems, Inc (Austin, TX) were assembled in pYES2 plasmid vectors. Sequences included the ADH1 glucose promoter with Genbank accession number U33753.1 involved in sugar metabolism, the SNF3 glucose receptor with Genbank accession number XM_001386379.1 (a high affinity glucose receptor already proven as a glucose sensor), ribosomal switch TC aptamer with Genbank accession number d26134.1, and Cyan reporter protein Genbank accession number KT878729.1 [23]. The experimental laboratory procedures followed the standard methods of molecular biology and synthetic biology [7,24-26]. The glucose sensors were constructed by site-directed cloning with specific primers (Figure 2), as follows:

Figure 1. Since both ends of assembled genes were flanked with EcoR1 and NotI sites, glucose device was digested with same enzymes to cut out insert from pYES2 vector. 4819 Fragment represents assembled glucose device, whereas, 5829bp fragment is pYES2 vector.
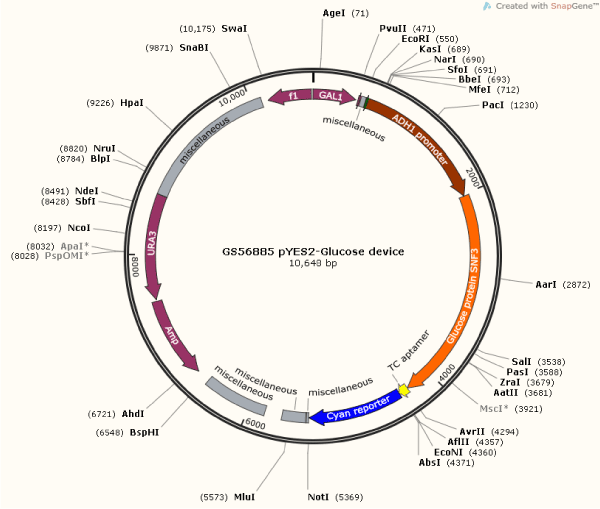
Figure 2. Complete Map of Glucose DNA Sensor Device. Illustration showing constructed plasmid the direction, placement and size of genes, and the respected enzymes within the pYES2 vector. Plasmid illustrates the glucose sensor beginning with GAL 1promoter, T7 promoter, riboswitch TC aptamer, ADH1 promoter, RBS, SNF3 protein, and fluorescent reporter protein sequences.
Assembly of Glucose Device: Five genes were assembled according to the designed device by the International Park of Creativity (IPOC). The assembled genes include: 1. T7 Promoter 2. ADH1 Promoter 3. SNF3 4. TC Aptamer Ribosomal-switch 5. Cyan Reporter. PCR primers corresponding to overlap sequences of each gene were used; the PCR reagents used were from New England Biolab. The pYES2 Vector used was from Invitrogen Inc. (CA).
Procedure: This procedure was carried-out by combining standard experimental laboratory methods of molecular biology and synthetic biology [7,24-26] with laboratory methods from Clonetex Systems, Inc (Austin, TX). Thus, PCR amplification was done as follows: Each gene was PCR amplified with gene-specific overlap primers with added restriction sites where needed. Both ends of assembled genes are flanked with an added EcoR1 site at 5’ and NotI site at 3’ so that fully assembled device can be sub-cloned into pYES2 vector.
PCR amplified pieces of all fragments were combined by using homologous recombination and sub-cloned into pYES2 vector. Clones obtained after transformation were sequenced and analyzed for correct construct.
Design of Glucose Device: This device contains five genes that are arranged in following order:

At the completion of PCR of all 5 targeted genes, homologous recombination of Glucose Device and sub-cloning into pYES2 vector, 4 clones were selected from transformed plate and processed for full length DNA sequencing. Clone with 100% matching sequence corresponding to the construct design was selected and purified to obtain plasmid construct at mid-scale purification level. This DNA device will allow us to detect proteins related to glucose presence in patient blood samples, and/or to detect existence of glucose itself.
Transformation of Yeast Saccharomyces cerevisiae (Sc): All assembled cassette parts which were amplified by PCR (pUC13 forward and reverse primers) and inserted into pYES2 plasmid were transformed into S. cerevisiae competent cells following standard method in combination with Invitrogen kit protocol (Invitrogen Inc., CA) [25,26]. However, the Invitrogen Inc. protocol was preferred in the denoted investigation as follows:
• Ten ml of YPD medium was inoculated with colonies of INVSc1, and incubated at 30oC under shake conditions, overnight.
• Optical Density (OD) of the yeast culture was determined at 600 nm, and then diluted in YPD medium, and incubated at 30°C for 2-4 hours.
• Culture was centrifuged at 2500 RPM, and pellet was obtained and it was resuspended in 40 ml in 1X TE buffer.
• Pellet was resuspended in 2 ml of 1X LiAc/0.5X TE buffer.
• Cells were incubated at room temperature for 10 minutes.
• For each transformation, 1 µg of plasmid DNA was mixed into 100 µg denatured sheared salmon sperm DNA, and it was also mixed with 100 µl of the yeast suspension from Step 5.
The plasmid solution was subjected to a series of reagents including buffer mixtures. The final solutions were subjected to centrifugation in a micro-centrifuge, and supernatant was removed, and the cell pellets were resuspended in 1X TE buffer and repellet. The cell pellet was resuspened in 50-100 µl 1X TE buffer and plated on a selective plate. Clones holding genetic devices were selected in Sc–URA plates.
Protein Analysis
In a different experiment, wild type Saccharomyces cerevisiae (Sc) strain 200892 was purchased from ATCC. 100 μL aliquots of the initial cell suspension were resuspended in YPD liquid broth (Sigma- Aldrich: 10 g of yeast extract, 20 g of peptone, 20 g of glucose and 18 g of agar for plates per liter) and grown for 6 h at 30°C reaching an optical density (OD) at 600 nm (λ 600) of 1. Standard synthetic complete medium, SC – URA (1.7 g of yeast nitrogen base, 5 g of ammonium sulfate, 2 g of – URA dropout mix, 5 ml of 200x leucine and 18 g of Difco agarL-1) supplemented with arginine (80 mgL-1; Sigma-Aldrich), and histidine (40 mgL-1; Sigma-Aldrich) was prepared for selection and culture of transformed yeast. The following protocols were implemented by Clonetex Systems, Inc. (Austin, TX), in order to identify proteins involved in detecting glucose. Samples of yeast cultures including transfected yeast with different genetic parts for detecting glucose and samples of non-transfected yeast (control) were subjected to protein extraction as follows. Each was mix with phosphate-buffered saline and centrifuged 3 times at 4°C at 2000 g for 300 s. The pellets were collected and 1g of pellet was used for subsequent protein analysis by two-dimensional difference gel electrophoresis (2D DIGE) and protein identification by liquid chromatography tandem mass spectrometry (LC-MS/MS) [8,27].
The Y-PER plus, dialyzable yeast protein extraction reagent was used to lyse yeast cells. The manufacturer’s protocol was followed; protease inhibitory cocktail and 0.1m dtt were used to enhance lysis of stationary phase S. cerevisiae.
The basic sample buffer (Sigma-Aldrich: 7m urea, 2m thiourea, 4% wt/vol chaps, and 30 mm tris pH 9) was added to the pellets. Following this, samples were purified using the 2d cleanup kit and the 2d quant kit (both from GE).
The protein samples were first tested on small 11cm 2d criterion gels (Biorad); proteins were visualized using silver staining. This step allowed determination of sample cleanliness and optimization of buffer pH range and percentage of acrylamide to use in gels in subsequent analytical steps.
Isoelectric focusing strips (GE life sciences) were used according to the manufacturer’s protocol to accomplish separation of proteins by 2D-DIGE. Protein spots were then processed for identification as follows. Tandem mass spectrometry coupled to liquid chromatography (LC-MS/MS): excised protein spots from the 2D-DIGE gel were digested in trypsin (10 μg ml-1; Sigma-Aldrich) at 37°C overnight [29]. Analyses of protein spots were carried out using a thermofinnigan ltq linear quadrupole ion trap mass spectrometer (san jose, ca) equipped with a michrom paradigm MS4 HPLC, a spectrasystems as 3000 auto sampler, and a nanoelectrospray source. Peptides were eluted from a 15 cm pulled tip capillary column (100 μm i.d. X 360 μm o.d; 3-5 μm tip opening) packed with 7 cm vydac c18 (Hesperia, ca) material (5 μm, 300 x 10-10 m pore size), using a gradient of 0-65% vol/vol solvent b (98% vol/vol methanol/ 2% vol/vol water/ 0.5% vol/vol formic acid/ 0.01% vol/vol triflouroacetic acid) over a 3600 s period at a flow rate of 350 nL min-1. The ltq electrospray positive mode voltage was set at 1.6 kv, and the capillary temperature was 180°C. Dependent data scanning was performed by the xcalibur v 1.4 software with a default charge of 2, an isolation width of 2.491 x 10-24 g, an activation amplitude of 35%, an activation time of 30 ms, and a minimal signal of 100 ion counts [30]. Global dependent data settings were as follows: reject mass width of 2.491 x 10-24 g, dynamic exclusion enabled, exclusion mass width of 2.491 x 10-24 g, repeat count of 1, repeat duration of 60s, and exclusion duration of 300s. Scan event series included one full scan with mass range 5.811 x 10-22 – 3.321 x 10-21 g followed by 3 dependent ms/ms scans of the most intense ion. Tandem ms spectra of peptides were analyzed with turbosequest v 3.1, a program that allows the correlation of experimental tandem MS data with theoretical spectra generated from known protein sequences [31]. The criteria that were used for a preliminary positive peptide identification are the same as previously described, namely peptide precursor ions with a +1 charge having an xcorr >1.8, +2 xcorr > 2.5 and +3 xcorr > 3.5. A dcn score > 0.08 and a fragment ion ratio of experimental/theatrical >50% were also used as filtering criteria for reliable matched peptide identification [30]. All matched peptides were confirmed by visual examination of the spectra. All spectra were searched against an S. cerevisiae database downloaded from NCBI in august 2010. At the time of the search, the yeast protein database from NCBI contained 72,216 entries. The results were also validated using xtandem, another search engine and with scaffold, a program that relies on various search engine results (i.e., : sequest, xtandem, mascot) and which uses bayesian statistics to reliably identify more spectra [30-32]. analytical 2D-DIGE analysis of samples to establish pH/pi range: two dimensional difference gel electrophoresis incorporating a pooled internal standard was performed. Four replicates for each experiment were performed. All data were analyzed in the program sigmaplot, using the shapiro-wilk test, and one-way anova in order to compare treatments and analyze correlations.
In vitro analysis of the glucose DNA sensor in whole blood samples from patients
Standard molecular biology methods were used to perform In vitro analysis of DNA of the glucose sensor in whole blood samples from patients in combination with the DNA extraction kit protocol Thermal Fisher (Grand Island, NE) [26]. PCR was performed in standard 5332 eppendorf thermocycler. DNA was quantified using a ge nanovue spectrophotometer; DNA was visualized and purified via electrophoresis using a thermo ec (ec-150) power supply. Gel visualization, and concentrations of both glycaemia and DNA of the glucose sensor were used as parameters to measure In vitro the expression of the DNA device.
Fluorescence (FSU) of the yeast DNA glucose sensor was determined In vitro. This was carried-out by growing the yeast DNA glucose sensor in culture media. Samples from the yeast DNA glucose device kept at -80°C were taken into YPD media and culture dilutions were made until they reached an OD at 600 nm of 1, cells were grown and sub-cultured in sterile-deionized water with different glucose blood equivalent concentrations (from 0 - 500 mg dL-1) for different time intervals ranging from 1 hour to 48 hours. Although different ratio of yeast DNA glucose sensor : sterilized-deionized water were used (i.e., 1:1, 2:1, 3:1), only the ratio 3:1 showed the most efficacy. The fluorescence (FSU) of the yeast DNA glucose sensor at this ratio was 1900 FSU. Therefore, when the yeast DNA glucose sensor was used in mixture with blood plasma samples for detection of glucose, this value (1900 FSU) was subtracted from the In vivo glucose testing results, as it is expressed in In vivo essay below. Fluorescence (FSU) of the DNA glucose sensor was measured in a GloMax Bioanalyzer Detector, GloMax®-Multi+ Detection System with Instinct™ Software (Promega, Madison, WI).
In vivo clinical glycaemia determinations in blood plasma from patients
The efficacy of the sensor was assayed In vivo, and fluorescence (FSU) was correlated with the concentrations of glycaemia in blood plasma samples from patients. This glycaemia determination was carried out at a clinical laboratory (Laboratory Alvarez-Medina) in Cali, Colombia. Blood plasma samples from patients was used for clinical glycaemia testing. Blood plasma was extracted by standard centrifugation method, thus, blood samples were taken from patients 8 hours prior to breakfast (fasting). Ten mL of blood sample was used and kept at room temperature (25-28°C) for 20 minutes. Then, samples were subjected to centrifugation at 3000 RPM for 10 minutes, in order to separate the plasma which will be used for testing. Glucose analysis was done in blood plasma samples from patients, following the American Diabetes Association (ADA) protocol. The method used was based in using Glucose-Oxidase/Peroxidase-Hexokinase test. One mL of reagent and 10 µL of blood plasma sample were mixed, and then incubated at 37°C for 10 minutes. The glycemic level was determined by using BTS 310 A at 540-546 nm wavelength.
Glycemic levels were classified intro 3 groups as follows:
1. 140 mg/dL or less = Normal
2. 140 mg/dL – 199 mg/dL = Pre-diabetes
3. 200 mg/dL or higher = Diabetes
In vivo fluorescence detection of DNA glucose sensor in blood plasma sample from patients
The determinations were performed as follows: blood plasma samples were used from 3 different groups of patients; normal, pre-diabetic, and diabetic at a clinical laboratory in Cali, Colombia as described above. Samples from 23 patients were used for analysis. These samples were mixed with the yeast DNA glucose sensor, at different ratio, and subjected to vertex for 30-50 seconds. Although different ratio of the yeast DNA glucose sensor : blood plasma were used (i.e., 1:1, 2:1, 3:1), only the ratio 3:1 showed the most efficacy. A total sample volume of 250 µL was used in a ratio of 3:1 (170 µL DNA glucose sensor : 80 µL blood plasma sample) for fluorescence detection of glucose. The natural inherent fluorescence (1900 FSU) of the yeast was subtracted from the final fluorescence of each sample. The results were then divided in 3 groups according to the intensity of the fluorescence in relation to the clinical glycaemia concentration, as described above. Therefore, the results of the fluorescence of the DNA glucose sensor were also divided into 3 groups in relation to glycaemia groups (i.e., normal, pre-diabetic, and diabetic). Thus, these groups were based on the mean from ranges of fluorescence intensity. Although, samples were collected from 23 patients, only 15 are shown in the denoted investigation.
Results
Construction of DNA Glucose Sensor.
Effective assembly of the DNA sensors for glucose sensor (Figure 2) was confirmed by the high fluorescence intensity (i.e., 1900 FSU) of the yeast DNA glucose sensor when exposed to YPD glucose media, as compared to a DNA glucose sensor grown in a media without glucose, which showed minimal fluorescence (<50 FSU). The assembly of the construct DNA glucose sensor was also confirmed by PCR analysis, which showed full assembly of the different genetic parts (Figure 1). Then, this DNA glucose sensor was sub cloned into pYES2 vector, and the device was effectively hosted in INVSc1 competent cells, as illustrated in the map (Figure 2).
Protein Analysis
After exposure to different concentrations of glucose including 411 mg/dL, and evaluation by 2D-DIGE and HPLC-MS/MS, the glucose DNA sensor over-expressed different proteins (Figure 4 and 5). Different spots of each representing increased or decreased amount of protein were analyzed and identified. Spots represent the proteins that were selected among many expressed, using laser bean (Figure 3). Minimum of 6 identified proteins expression was increased (i.e., pyruvate kinase1, phosphomanno mutase, protien disulfide isomerase, and others), while expression of at least 6 were identified as decreased (i.e., glycinamide ribonucleotide synthetase, thioredoxin reductase, glutamate synthase, and others). For example, when the glucose DNA sensor was exposed to glucose, proteins related to metabolism (i.e., pyruvate kinase 1, s-adenosylmethionine synthetase 2, glycinamide ribonucleotide synthetase, phosphomannomutase, and protein disulfide isomerase) and regulation of glucose exhibited higher expression levels. Up regulation of these proteins was significantly different from control or non-transformed yeast cells in which proteins involved in glucose sensing were down regulated or were not highly expressed.

Figure 3. 2D-DIGE Gel of Proteins of Glucose DNA Sensor Two dimensional (2D-DIGE) gel showing fluorescence of the proteins of our DNA sensor at different intensities. The brightest spots indicating higher protein expression were selected for analysis under HPLC mass spectrometry and laser identification.
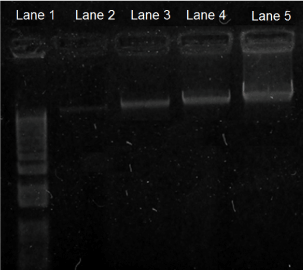
Figure 4. Electrophoresis Bands of DNA of the Glucose Sensor after Exposure to Blood Serum Samples from Patients. Electrophoresis gel bands of DNA of the glucose sensor, expressing different fluorescence after exposure to blood samples from different patients (In vitro test). DNA gel bands from glucose sensor as shown from left to right: Lane 1. Molecular DNA marker (1 kb). Lane 2. Normal patient sample; this gel showed the least brightness, and the smallest concentration of both glycaemia (100.2 mg/dL) and DNA (18.5 ng). Lane 3. Pre-diabetic patient sample; this gel showed the second least brightness, and second smallest concentration of both glycaemia (135.9 mg/dL) and DNA (52.5 ng). Lane 4. Controlled diabetic patient sample; this gel showed the second highest brightness, and the second highest concentration of both glycaemia (185.7 mg/dL) and DNA (77 ng). Lane 5. Uncontrolled diabetic patient sample; this gel showed the highest brightness, and the highest concentration of both glycaemia (411 mg/dL) and DNA (85 ng).

Figure 5. Hypothetical mechanism of DNA sensor for detection of glucose for Diabetes. This mechanism is based on the following steps (#1 to #6): #1 = Yeast Cell. #2 = Glucose molecule in contact with yeast cell. #3 = Ion channel in the cell membrane. Ion channel excited by glucose molecule. #4 = Second messengers system for intracellular glucose transport (i.e. GLUT and Kinases) in the cytoplasm (Olson and Pessin, 1996). Induction of the metabolic cascade toward activation of the DNA sensor in the nucleus. #5 = Transcription of DNA sensor in the nucleus toward production of the fluorescence reporter protein. #6 =Excitation of bio-photons for emission of fluorescence according to glucose concentration. (46)
In vitro analysis of the glucose DNA sensor in whole blood samples from patients
Electrophoresis gel bands of DNA of the glucose sensor showed different fluorescence after exposure to blood samples from patients with different levels of glycaemia (i.e., normal, pre-diabetic, diabetic). Thus, DNA gel bands from the glucose sensor showed different intensities depending upon the level of glycaemia of the patient. Hence, the brightness levels correlate with both concentrations of DNA and levels of glycaemia, samples from normal patients expressed the least brightness, and the smallest concentrations of both DNA (18.5 ng) and glycaemia (100.2 mg/dL); samples from pre-diabetic patients expressed the second least brightness, and the second smallest concentrations of both DNA (52.5 ng) and glycaemia (135.9 mg/dL); samples from controlled diabetic patients expressed the second highest brightness, and the second highest concentrations of both DNA (77 ng) and glycaemia (185.7 mg/dL); and samples from uncontrolled diabetic patients express the highest brightness, and the highest concentrations of both DNA (85 ng) and glycaemia (411 mg/dL) (Figure 4).
In vivo clinical glycaemia determinations in blood plasma from patients
Our In vivo results relate to the In vitro results reported above (Figure 4). Although, samples were collected from 23 patients, only results from 15 patients are shown in the denoted investigation. Results are based on the means of the range of the fluorescence intensity per each group (i.e. group 1 = normal, group 2 = pre-diabetic, group 3 = diabetic) (Table 1). There was a correlation between fluorescence intensity emitted by the DNA glucose sensor when mixed with blood plasma from patients, and the level of glycaemia clinically tested in patients. Thus, the fluorescence means of samples from different groups according to the range of fluorescence intensity (i.e., group 1 (normal), group 2 (pre-diabetic), and group 3 (diabetic)) correlates with an equivalent level of glycaemia. These correlations are shown in Table 1, hence, the mean of the fluorescence from the DNA glucose sensor exposed to blood plasma samples from normal patients showed the lowest intensity (1068.8 FSU ± 141.4), and the lowest level of glycaemia (<140 mg/dL); the mean of samples from pre-diabetic patients showed the second highest intensity (4783.3 FSU ± 144.3), and the second highest level of glycaemia (140-199 mg/dL); and the mean of samples from diabetic patients showed the highest intensity (6712 FSU ± 1118), and the highest level of glycaemia (>200 mg/dL).
Table 1: Group Types of Patients According to Fluorescence of Glucose DNA Sensor and Clinical Glycemia Concentration.
|
Patient |
Gender
(Female/Male) |
Age |
Glucose Fluorescence (FSU) |
Glycemia (mg/dL) |
Group 1
(Normal) |
19 |
Female |
40 |
1000 |
104 |
18 |
Female |
34 |
940 |
106 |
16 |
Male |
64 |
1000 |
108 |
15 |
Female |
60 |
1100 |
110 |
17 |
Female |
67 |
1000 |
110 |
10 |
Male |
46 |
960 |
130 |
11 |
Female |
62 |
1350 |
135 |
7 |
Female |
60 |
1200 |
138 |
|
|
|
|
Mean = 1068.8 SD = ± 141.4 |
|
|
|
|
|
|
|
Group 2
(Pre-Diabetic) |
23 |
Male |
76 |
4700 |
151 |
8 |
Male |
52 |
4700 |
190 |
22 |
Female |
66 |
4950 |
196 |
|
|
|
|
Mean = 4783.3 SD = ± 144.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Group 3
(Diabetic) |
21 |
Male |
65 |
7400 |
218 |
13 |
Male |
42 |
7500 |
243 |
21 |
Male |
51 |
7500 |
346 |
12 |
Male |
56 |
5200 |
370 |
|
|
|
|
Mean = 6712 SD = ± 1118 |
|
*Note ± SD = Standart Deviation
*Group Types of Patients = Normal, Pre-Diabetic, and Diabetic
On the contrary, no fluorescence was exhibited by negative control which was a non-transformed S. cerevicea. Therefore, the glucose yeast DNA sensor showed higher levels of fluorescence for both higher and lower concentrations of glucose, as compared to control or non-transformed yeast cells.
Discussion
The assemblage of standardized biobrick parts with synthesized sequences such as ADH1 protein, SNF3 transporter, ribosomal switch TC aptamer, and fluorescence reporter proteins, was achieved effectively, as reflected in the sensitivity of our glucose yeast DNA sensor to glucose In vitro and In vivo in human blood plasma samples. The results confirm the efficacy of the device in detecting proteins related to glucose presence, which is commonly found in eukaryotic glycoproteins, and/or detecting glucose itself. Other reports have shown glucose sugar associated with human glycoproteins [7].
Clear evidence of the functionality of our yeast DNA glucose sensor is observed in the In vitro direct correlation between the fluorescence of the DNA glucose sensor and in the concentration of glucose, and by the direct correlation between growth of yeast cells carrying the sensor, glucose concentration, and fluorescence (data not shown). Additionally, the results from our investigation showed direct correlation between concentrations of glycaemia and DNA, confirming the accuracy of the DNA sensor. These results also demonstrate the specificity of the DNA sensor for diabetic patients. Previous reports have also demonstrated the direct correlation between fluorescence of DNA sensor cells and sensitivity of detection [7].
Likewise, the efficacy of S. cerevisiae as an ideal competent cell (chassis) for glucose sensors has been demonstrated, due to this yeast’s vast genetic machinery related to sugar metabolism [22]. Further, the addition of genetic parts such as ADH1 protein, SNF3 transporter, and ribosomal switch TC aptamer, enhances the efficiency and ability of S. cerevisiae to detect different or specific concentrations of glucose [7]. ADH1 protein and the SNF3 transporter of glucose, along with the ribosomal switch TC aptamer, enhance the sensitivity of the sensors, even for very low levels of glucose. Shall we keep in mind that the Eukaryote S. cerevisiae cells may carry their own native ribosomal switch. Additionally, selected promoters under controlled carbon catabolism repression enhance the detection of glucose by the DNA sensors. Thus, sensor proteins detect glucose molecules in the blood when the sensor comes into contact with a sample. The initial response depends on glucose concentration. Once the promoter activates transcription, depending on glucose concentration, certain levels of transcript are translated into protein. The ribosomal switch aptamer ensures transcription and translation of proteins when high levels of glucose might otherwise repress activation of ADH1 protein. When the ribosomal switch TC aptamer is constitutively on, protein expression increases [7] (Figure 2). The genes for transcribed proteins are fused to genes for a protein that fluoresces according to its expression, which correlates directly with glucose concentrations in blood plasma samples. Other authors have suggested repression of the operon containing genes for ribosomal proteins when the amount of free proteins exceeds the amount of ribosomal proteins, thus creating an on/off switch system [36,37]. A riboswitch has also been identified as a signaling mechanism in response to extreme environmental conditions such as oxidative stress [38]. Thus, riboswitches may play an important role in the DNA sensor described herein, since glucose is susceptible to oxidative conditions [39].
Therefore, we are herein suggesting a hypothetical mechanism of interaction between the DNA glucose sensor and glucose molecules (Figure 5). This mechanism is based on the following steps: yeast DNA glucose sensor cells become in contact with glucose molecule, ion channel in the yeast cell membrane becomes activated, then second messengers system for intracellular glucose transport (i.e. GLUT and Kinases) in the cytoplasm induce the metabolic cascade toward activation of the DNA sensor in the nucleus, following transcription of DNA sensor in the nucleus towards production of the fluorescence reporter protein, and finally the excitation of bio-photons for emission of fluorescence according to glucose concentration occurs [40-42].
2D-DIGE and LC-MS/MS analysis have demonstrated that our genetic glucose sensor device is able to induce a differential response in a yeast DNA sensor, compared to a non-transformed yeast control, even when both yeast systems are grown under similar conditions and similar glucose concentrations. Proteins involved in, or related to, glucose metabolism were expressed at high levels. Up-regulation of these proteins was demonstrated by the glucose yeast DNA sensor as seen in Figure 3, and was highly correlated to glucose metabolism, indicating that these proteins might also have aided the detection of glucose by the DNA sensor through their support of the translation of yeast proteins and amino acid biosynthesis [43,44]. Proteins that were highly expressed in our DNA sensor included s-adenosylmethionine synthetase 2, glycinamide ribonucleotide synthetase, phosphomannomutase, and protein disulfide isomerase [41,45]. Also, these proteins may, in turn, regulate the expression of the sensor device proteins. Some of the up-regulated proteins are involved in the regulation of sensor device protein expression, including glutamate synthase and translationally-controlled tumor protein (TCTP) analog, both of which have an inhibitory effect on down-regulation of the enzymes of glycolysis [5]. The high sensitivity of 2D-DIGE and HPLC-MS/MS techniques also ensured the correct identification of proteins of interest, since many conventional molecular biology techniques are not sensitive enough to identify these proteins at different regulation states [21].
The expression of the fluorescence by the DNA glucose sensor in each group (i.e., normal, pre-diabetic, and diabetic) which correlated well with the glycaemia clinical test, corroborates the accurate sensitivity of the detection of the sensor when using the optical GloMax system. Perehaps, this is explained by the active optical behavior of living cells in response to photon emition induced by external source of luminescence [46]. This optical photon activity resembles auto-fluorescence which might vary along the time. Similarly, biological systems, including organisms, might follow this variable pattern until the level of spontaneous optical activity is recoverd [47]. Thus, organisms and/or cells can be excited by a light-photon source with a specific spectrum and intensity. Therefore, it seems that living organisms are under the constant effect of wide spectral range of photons. Thus, it is more likely that a form of EMF emission is also involved in the fluorescence of the biological system, such as the DNA glucose sensor [1].
Conclusion
Our DNA sensor exhibited a high degree of sensitivity and was capable of detecting both, very low levels and high levels of glucose, equivalent to the clinical glycaemia values. This attribute of the DNA sensor is highly advantageous, since detecting lower levels of sugar in blood will permit clinicians to diagnose pre-diabetes and/or hypoglycemia in a subject. Since our DNA sensor allows for the detection of both high and low levels of glucose, this one device may be used for multiple purposes, thus making our DNA sensor extremely cost-effective.
Constructed DNA sensor2021 Copyright OAT. All rights reservdized genetic parts and were able to detect low and/or high levels of glucose In vitro and In vivo in human blood plasma samples in a short period of time (i.e., less than a minute).
This cost-effective DNA sensor is good since our sensor can be used for early diagnosis of diabetes or pre-diabetic conditions, thereby allowing for earlier clinical intervention. The direct correlation between levels of glucose and the intensity of fluorescence of the DNA sensor shows the advantage of using this technology in order to identify specific types of diabetic patients, since sometimes it is difficult to establish accurate correlations with different methods.
Acknowledgement
This work was supported by the International Park of Creativity laboratory, Manizales, Colombia. We thank Clonetex Systems Inc, Austin, TX, for providing the synthesized sequences, and technical assistance during the implementation of this work. Also, to Dr. Cecilia Arturo, M.D. for her contribution with medical advice and providing patients, and to Dr. Lawrence Villanueva for editing the manuscript.
References
- Cifra M, Fields JZ, Farhadi A (2011) Electromagnetic cellular interactions. Prog Biophys Mol Biol, 105: 223-246. [Crossref]
- D’Souza SF (2001) Microbial Biosensor. Review. Biosensors, and Bioelectric. Elsevier, 337-353.
- Cook DJ, Holder LB (2011) Sensor Selection to Support Practical Use of Health-Monitoring Smart Environments. Data Min knowl Discov 1: 339-351. [Crossref]
- Hahm JI (2011) Functional Polymers in Protein Detection Platforms: Optical, Electrochemical, Electrical, Mass-Sensitive, and Magnetic Biosensors. Sensors 11: 3327-3355.
- Liu X, Germaine KJ, Ryan D, Dowling DN (2010) Genetically modified Pseudomonas biosensing biodegraders to detect PCB and chlorobenzoate bioavailability and biodegradation in contaminated soils. Bioeng Bugs 1: 198-206. [Crossref]
- Rusling JF, Kumar CV, Gutkind JS, Patel V (2010) Measurement of biomarker proteins for point-of-care early detection and monitoring of cancer. Analyst 135: 2496-2511. [Crossref]
- Cuero R, Lilly J, McKay DS (2012) Constructed molecular sensor to enhance metal detection by bacterial ribosomal switch-ion channel protein interaction. J of Biotechnol 158: 1-7. [Crossref]
- Murakami T, Nishiyama T, Shirotani T, Shinohara Y, Kan M, et al. (1992) Identification of two enhancer elements in the gene encoding the type 1 glucose transporter from the mouse which are responsive to serum, growth factor, and oncogenes. Journal of Biological Chemistry 267: 9300-9306.
- Cuero R, McKay (2013) Induction and construct UV protective yeast plasmid. J Biotechnology 166: 76-83. [Crossref]
- American Diabetes Association (2002) Screening for diabetes. Diabetes care 25(1): S21-S24.
- Gillett MJ (2009) International Expert committee report on the role of the A1c assay in the diagnosis of diabetes. Diabetes care 32, 1327-1334. Clin Biochem Rev 30: 197-200. [Crossref]
- Jacquez JA (1984) Red blood cell as glucose carrier: significance for placental and cerebral glucose transfer, Am J Physiol 246: R289-98. [Crossref]
- Cramp DG (1967) New automated method for measuring glucose by glucose oxidase. J Clin Pathol 20: 910-912. [Crossref]
- Fritsche HA, Dee JW, Adams-Park HR (1975) Enzymatic endpoint analysis of glucose with the hexokinase method and the Union Carbide fast centrifugal analyzer. Clin Biochem 8: 240-6. [Crossref]
- Bruckner A (1980) The kinetic determination of glucose with the glucose dehydrogenase method using the Eppendorf automatic analyzer 5040 (author's transl). J Clin Chem Clin Biochem 18: 275-278. [Crossref]
- Kulys J, Tetianec L, Bratkovskaja I (2010) Pyrroloquinoline quinone-dependent carbohydrate dehydrogenase: activity enhancement and the role of artificial electron acceptors. Biotechnol J 5: 822-8. [Crossref]
- Frias JP, Lim CG, Ellison JM, Montandon CM (2010) Review of adverse events associated with false glucose readings measured by GDH-PQQ-based glucose test strips in the presence of interfering sugars. Diabetes Care 33: 728-9. [Crossref]
- Kelly BN, Haverstick DM, Bruns DE (2010) Interference in a glucose dehydrogenase-based glucose meter. Clin Chem 56: 1038-40. [Crossref]
- Weiss JN (1991) Diabetes detection method (United States Patent 5025785).
- Mackay IR, Rowley MJ, Zimmet PZ (1998) Methods for the diagnosis of diabetes and prediabetic conditions (United States Patent 5770381).
- Chang CC, Huang CN, Chuang LM (1998) Autoantibodies to thyroid peroxidase in patients with type 1 diabetes in Taiwan. Eur J Endocrinol 139: 44-8. [Crossref]
- Cuero R, Ouellet T (2005) Metal ions modulate gene expression and accumulation of the mycotoxins aflatoxin and zearalenone. J Appl Microbiol 98: 598-605. [Crossref]
- Brown V, Sexton JA, Johnston M (2006) A glucose sensor in Candida albicans. Eukaryot Cell 5: 1726-37. [Crossref]
- Davis LG, Kuehl WM, Battey JF (1995) Basic Methods in Molecular Biology, second ed. McGraw-Hill Professional.
- Gietz RD, Schiestl RH (2007) Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 35-7. [Crossref]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning laboratory manual. Second ed. Cold Spring Harbor Laboratory Press, New York.
- Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, et al. (2003) A novel experimental design for comparative two-dimensional gel analysis: two- dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics 3: 36-44.
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem 68: 850-8. [Crossref]
- Andon NL, Hollingworth S, Koller A, Greenland AJ, Yates JR, et al. (2002) Proteomic characterization of wheat amyloplasts using identification of proteins by tandem mass spectrometry. Proteomics 2: 1156-68. [Crossref]
- Cooper B, Eckert D, Andon NL, Yates JR, Haynes PA (2003) Investigative proteomics: identification of an unknown plant virus from infected plants using mass spectrometry. J Am Soc Mass Spectrom 14: 736-41. [Crossref]
- Craig R, Beavis RC (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20: 1466-1467. [Crossref]
- Eng JK, McCormack AL, Yates III JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976-89. [Crossref]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383-92. [Crossref]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75(17): 4646-58. [Crossref]
- Grundy FJ, Henkin TM (2006) From Ribosome to Riboswitch: control of gene expression in bacteria by RNA structural rearrangements. Crit Rev Biochem Mol Biol 41: 329-38. [Crossref]
- Barham D and Trinder P (1972) An improved colour reagent for the determination of blood glucose by oxidase system. Analyst 97: 142-5. [Crossref]
- Vitreschak AG, Rodionov DA, Mironov AA, Gelfang MS (2004) Riboswitches: the oldest mechanism for the regulation of gene expression. Trends Genet 20: 44-50. [Crossref]
- Bommer UA and Thiele BJ (2004) The translationally controlled tumour protein (TCTP). Int J Biochem Cell Biol 36: 379-85. [Crossref]
- Elliott P, Peakman TC (2008) The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int J epidemiol 37: 234-44. [Crossref]
- Im H, Wittenberg NJ, Lesuffleur A, Lindquist NC, Oh SH (2010) Membrane protein biosensing with plasmonic nanopore arrays and pore-spanning lipid membranes. Chem Sci 1: 688-696. [Crossref]
- Prieto B, Miguel D, Costa M, Coto D, Alverez FV (2010) New quantitative electrochemiluminescence method (ECLIA) for interleukin-6 (IL-6) measurement. Clin chem Lab Med 48: 835-8. [Crossref]
- Thakral S, Manhas P (2011) Fiber Optic Sensors Technology & Their applications, Int J Comm Tech 2: 126-128.
- Trinder P (1969) Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann Clin Biochem 6, 24-27.
- Guyton, Arthur C (1976) Textbook of Medical Physiology (5th ed.) Philadelphia: W.B. Saunders.
- Diagnosing Diabetes and Learning About Prediabetes (2016) American Diabetes Association, November 21, URL. - See more at: http://www.diabetes.org/diabetes-basics/diagnosis/
- Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 4th ed. Burtis CA, Ashwood ER, Bruns DE. WB Saunders Co, 2005.
- Olson AL, Pessin JE (1996) Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr, 16: 235-256.