Apoptosis is the major cause of death of neurons in various pathologic neurological conditions and in physiologic developmental death. The most common human neurodegenerative disease is Alzheimer’s disease (AD) which is also a major form of dementia. AD is characterized by senile plaques comprising Amyloid-β (Aβ), neurofibrillary tangles comprising hyper phosphorylated tau proteins and Neurodegeneration [1]. Aβ is aberrantly produced in AD patient’s brain and soluble oligomeric Aβ is the prime pathological species since it induces synapse pathology [2,3] and apoptosis of both neurons [4,5] and astrocytes [6]. Lack of neurotrophins such as Nerve Growth Factor (NGF) play a major role in the development of various neurological conditions. In fact, it has been reported that lack of NGF signaling leads to sporadic AD [7,8]. Neuron death is a physiologic process which occurs during brain development and it is noted that half of the neurons die due to lack of trophic support such as limited NGF supplies [9]. However, the molecules which mediate neuron death under the disease condition or during the embryonic development have not been examined in much detail till now.
BH3-only proteins (BCL-2 homology domain 3) of the BCL-2 family are the surveillants of cellular stress, orchestrating cell death via apoptosis in neurons. Both, extrinsic as well as intrinsic cell death signals are initiated by the BH3-only proteins of the BCL-2 family. The members of the BCL-2 family of proteins contain either one or up to four of the BCL-2 Homology (BH) domains. The members can be classified as pro-survival e.g. BCL-XL, BCL-W, MCL-1 and pro-apoptotic e.g. BAX, BAK (comprising of three BH domains) and BIM, PUMA, NOXA, BMF, HRK, BNIP (comprising of only BH3 domain, or the BH-3 only proteins). It is through these domains that a complex interplay of interactions of these proteins ultimately determines the fate of cells. The pro-survival proteins inhibit apoptosis by sequestering the pro-apoptotic proteins in healthy cells. The most crucial step in the propagation of the process of apoptosis is the interaction of the BH3-only proteins with the other Bcl-2 family members. In response to apoptotic stimuli such as DNA damage or growth factors deficiency, the BH3-only proteins are upregulated/activated which in turn bind to the pro-survival proteins thus relieving the death proteins, BAX and BAK [10]. BH3-only proteins promote apoptosis by two mechanisms; by direct activation of the pro-apoptotic proteins and by repressing the anti-apoptotic proteins. Some BH3-only proteins such as BAD, BMF, NOXA are selective to a subset of their pro-survival relatives, whereas other BH3-only proteins, BIM and PUMA can neutralize all the pro-survival proteins and can directly activate pro-apoptotic proteins such as BAX [11]. Our previous investigations reveal that pro-apoptotic BH3-only proteins BIM and PUMA play an essential role in mediating neuron death and axonal degeneration induced by NGF deprivation as well as Aβ insults [12-14]. However, other BH3-only proteins such as BMF may be required for displacement of BIM from pro-survival proteins for its full activity. It has been shown that displacement of BIM by BMF and PUMA from anti-apoptotic proteins mediates paclitaxel-induced apoptotic death of breast cancer cells [15].
Very recently we reported that BMF plays an essential role in neuron death evoked by neurotoxic Aβ oligomers or NGF deprivation [16]. We have shown for the first time, a co-operative role of BMF with BIM and PUMA in inducing neuron death in response to NGF deprivation or Aβ treatment. We investigated the mechanism in detail and our study revealed that upon exposure to Aβ or after NGF deprivation, there is a rapid increase in the expression of BMF in both transcript and protein levels in primary cultures of cortical neurons as well as neuronal PC12 cells. Examination of over-expression of BMF in neurons reveals that it causes increase in levels of (i) pH2AX, (ii) BAX, and (iii) cleaved caspase [3]. There are also decrease in mitochondrial membrane potential and number of neuronal cells. All these suggest that over-expression of BMF leads to apoptotic death of neuronal cells. Moreover, the loss of cortical and hippocampal neurons evoked by Aβ or sympathetic neurons after NGF withdrawal are abrogated by the down-regulation of BMF. Interestingly, our study reveals that death of neurons due to over-expression of BMF is blocked by knockdown of BIM and PUMA. These findings suggest that BMF induces apoptosis at least partially by activating BIM or PUMA or both. Lastly, down-regulation of these three BH3-only molecules (BIM, PUMA and BMF) by specific shRNAs provided enhanced protection to neurons against death evoked by Aβ neurotoxicity and NGF deprivation. Collectively, our study brings to light the crucial role of BMF in triggering neuronal death induced by Aβ toxicity and NGF deprivation at the molecular level. This highlights the novel finding of our study which includes the co-operative action of the three BH3-only proteins, (BIM, PUMA and BMF) in orchestrating the neuron death mechanism.
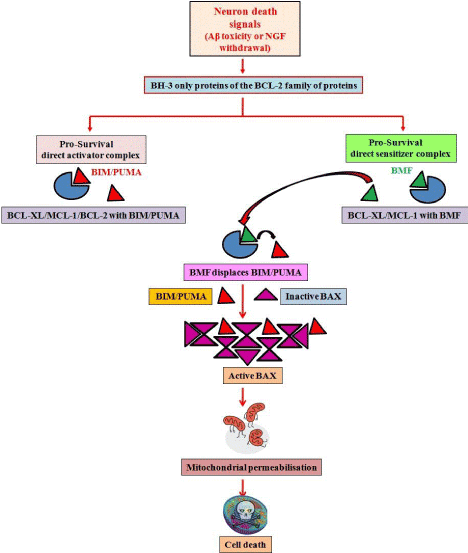
Our findings are corroborated by other recent investigations which further augment our conclusions. These recent reports also identify BMF as the major mediator of neuronal injury in response to glucose/oxygen deprivation [17]. Further, displacement studies have also been performed which propose the interaction of BMF with BIM, by way of which it sequesters the latter from other anti-apoptotic proteins, hence leading to the apoptotic cascade to function [15]. Our findings suggest a model (Figure 1) in which Aβ toxicity or NGF deprivation leads to expression/activation of BH-3 only proteins such as BIM, PUMA and BMF. BMF displaces BIM/PUMA from pro-survival proteins such as BCL-2, BCL-XL or MCL-1; subsequently BAX is activated by multimerization and leads to mitochondrial outer membrane permeabilization, caspase activation and cell death. Further studies on the interaction of BH-3 only proteins with other members of the family and cooperation among them in various death paradigms may reveal several undefined events in the molecular cascade of cell death.
- Murphy MP, LeVine H 3rd (2010) Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis 19: 311-323. [Crossref]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E (2010) Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol 119: 523-541. [Crossref]
- LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci 8: 499-509. [Crossref]
- Sanphui P, Biswas SC (2013) FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis 4: e625. [Crossref]
- Saleem S, Biswas SC (2017) Tribbles Pseudokinase 3 Induces Both Apoptosis and Autophagy in Amyloid-β-induced Neuronal Death. J Biol Chem 292: 2571-2585. [Crossref]
- Saha P, Biswas SC (2015) Amyloid-β induced astrocytosis and astrocyte death: Implication of FoxO3a-Bim-caspase3 death signaling. Mol Cell Neurosci 68: 203-211. [Crossref]
- Capsoni S, Cattaneo A (2006) On the molecular basis linking Nerve Growth Factor (NGF) to Alzheimer's disease. Cell Mol Neurobiol 26: 619-633. [Crossref]
- Cattaneo A1, Capsoni S, Paoletti F (2008) Towards noninvasive nerve growth factor therapies for Alzheimer's disease. J Alzheimers Dis 15: 255-283. [Crossref]
- Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453-501. [Crossref]
- Adams JM, Cory S (2018) The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ 25: 27-36. [Crossref]
- Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49-63. [Crossref]
- Akhter R, Sanphui P, Biswas SC (2014) The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in β-amyloid-induced neuron death. J Biol Chem 289: 10812-10822. [Crossref]
- Biswas SC, Shi Y, Sproul A, Greene LA (2007) Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J Biol Chem 282: 29368-29374. [Crossref]
- Akhter R, Sanphui P, Das H, Saha P, Biswas SC (2015) The regulation of p53 up-regulated modulator of apoptosis by JNK/c-Jun pathway in β-amyloid-induced neuron death. J Neurochem 134: 1091-1103. [Crossref]
- Kutuk O, Letai A (2010) Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ 17: 1624-1635. [Crossref]
- Akhter R, Saleem S, Saha A, Biswas SC (2018) The pro-apoptotic protein Bmf co-operates with Bim and Puma in neuron death induced by β-amyloid or NGF deprivation. Mol Cell Neurosci 88: 249-257. [Crossref]
- Pfeiffer S, Anilkumar U, Chen G, Ramirez-Peinado S, Galindo-Moreno J, et al. (2014) Analysis of BH3-only proteins upregulated in response to oxygen/glucose deprivation in cortical neurons identifies Bmf but not Noxa as potential mediator of neuronal injury. Cell Death Dis 5: e1456. [Crossref]
2021 Copyright OAT. All rights reserv