Creutzfeldt Jakob disease (CJD) is a highly fatal disease that presents with rapidly progressive dementia, with most patients dying within a year of onset. CJD presents a potential risk of iatrogenic transmission, as it can incubate asymptomatically in humans for decades before becoming clinically apparent. We present the case of a man with suspected CJD confirmed by biopsy and a review is made of the pathogenesis and clinical diagnosis of CJD.
Creutzfeldt Jakob desease, spongiform encephalopathy, prion protein, rapidly progressive dementia
CJD is considered the most common transmissible spongiform encephalopathy in humans, with 85% of reported cases being sporadic, while the remaining 15% correspond to the familial form of the disease and the iatrogenic form of transmission.
Any patient with rapidly progressive dementia associated with other signs of involvement of areas of the central nervous system, such as the presence of myoclonus, cerebellar disease, visual disorders, and / or psychiatric condition, among others, should be suspected in all patients. Combining clinical features with electroencephalogram, laboratory parameters, and neuroimaging findings will facilitate the diagnosis.
The case of a 61-year-old man, resident of Zamora, Michoacán, Mexico, dedicated to gardening is presented. History of systemic arterial hypertension.
His current condition began four months ago with anterograde amnesia, later hemiparesis in the left pelvic limb, vertigo and ataxia that leads to an increase in his plane of support.
He progressively evolved so he is referred to our hospital. He was admitted for study protocol due to probable brain neoplasia vs multiple sclerosis. The physical and neurological examination revealed an oriented and reactive patient, afebrile with isochoric and normoreflectic pupils. Without cranial nerve alterations. With the presence of monoparesis in the left pelvic limb, he was voluntarily discharged from the hospital.
Three months later, the patient was readmitted with worsening symptoms, the presence of rapidly progressive dementia, bedridden, with global aphasia and ataxia. Physical examination revealed an aphasic patient, with the presence of hyperreflexia in the pelvic limbs, however involuntary movements or myoclonus were not appreciated.
Magnetic resonance imaging was requested, which only evidenced the presence of pericalous calcifications. The diagnosis of Creutzfeldt Jacob's disease is suspected due to clinical symptoms and time of evolution, for which a brain biopsy is scheduled, tissue was sent to the pathology department. Fifteen days later, the histopathological diagnosis of spongiform encephalopathy is reported.
The term Creutzfeldt Jakob disease was used for the first time in 1922 by Spielmeyer when naming the condition described by two German physicians [1]. The 6 original subjects (1 case reported by Creutzfeldt and a series of 5 patients by Jakob) were a heterogeneous group of patients with unusual neuropathological findings associated with other disorders and years later only two of the cases were confirmed with modern techniques to what is currently Creutzfeldt Jakob disease. Forty years later (1960), the characteristic symptoms, the findings by electroencephalography, and the spongiform changes in the neuropil were recognized as the cardinal characteristics of the disease [2].
Creutzfeldt Jakob disease is included within the transmissible spongiform encephalopathies or prion diseases, which are a group of fatal neurodegenerative disorders in humans and animals. In addition to Creutzfeldt Jakob disease (CJD), prion diseases in humans include Gerstmann Sträussler Scheinker syndrome and fatal familial insomnia. The most common form of prion disease in humans is sporadic Creutzfeldt Jakob disease (CJD) with an incidence of 1 case per million people per year worldwide [3]. However, in 10-15% of cases it has a familial character (fCJD), this form is inherited with an autosomal dominant pattern (chromosome 20) [4].
In a small minority of patients, prion diseases are acquired via iatrogenic transmission (iCJD) (association with intracerebral electrodes, corneal transplantation, dura mater grafts and growth hormone injections, the first case report, in 1974, was in a patient who received a corneal transplant from an infected cadaver) [5]. As well as exposure to the agent of bovine spongiform encephalopathy (variant form of CJD (vCJD)) [4].
Pathogenesis
There have been two paradigm shifts in our understanding of transmissible spongiform encephalopathies (TSE) in the last 30 years. The first is the formulation, promotion, and subsequent general acceptance of the prion hypothesis as the best available explanation for TSE. The second (currently ongoing) is the extension of the prion paradigm to areas of normal cell physiology and the formulation of a general model for the mechanism involved in a wide variety of neurodegenerative diseases [6].
The normal cellular prion protein (PrPC) is a glycoprotein anchored to a glyphosphatidylinositol with a largely α-helical C-terminal domain and an intrinsically disordered N-terminal domain that binds to Cu2 + and Zn2 +. PrPC is typically exposed on the cell surface, but can also be located on the lumenal side of intracellular organelles or vesicles (Figure 1).
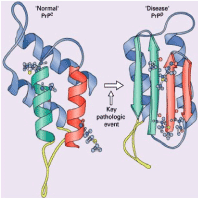
Figure 1. Prion protein tertiary structure. The normal cellular protein (PrPC) has an a-helical internal structure (coils). Conformational change into the pathogenic form (PrPSc) results in replacement by amyloidogenic anti-parallel b-pleated strands (arrows) [7].
PrPC is expressed in various cell types, both in the nervous system and in peripheral tissues, although it is more abundant in neurons. The vast majority of PrPC molecules are synthesized in the endoplasmic reticulum and in the Golgi apparatus as glycoproteins that bind to cell membranes via a glyphosphatidylinositol anchor. Typically, PrPC molecules follow the secretory pathway to the cell surface, where they are exposed to the extracellular environment. PrPC can then be re-internalized into endocytic vesicles and recycled to the cell surface. PrPC can also be internally cleaved at two different sites by endogenous proteins to generate N-terminal and C-terminal fragments.
Although it is clear that the conversion of PrPC to pathological forms underpins prion diseases, describing a general physiological role for PrPC in healthy individuals has been difficult. Manipulation of PrPC levels has been reported to influence a variety of cellular functions and result in altered host phenotypes, including alterations in metal homeostasis, development of synaptic plasticity, circadian rhythm, and stress responses [4,8].
In prion diseases, most, if not all, of the α-helical structure of PrPC folds into β-sheets. In addition to its involvement in prion diseases, one of the most recognized pathophysiological roles of PrPC is the mediation of some of the neurotoxic effects of β-amyloid oligomers in models of Alzheimer's disease [9]. The prion hypothesis poses an epigenetic agent, composed largely, if not exclusively, of an altered, refolded, and aggregated host-encoded prion protein (PrPC) in the disease-associated form (termed PrPSc).
It is proposed that this conversion process is autocatalytic, PrPSc is synonymous with the infectious agent, and PrPSc production is the main event causing neurodegeneration. Within this paradigm, some of the more unusual features of TSEs become more understandable: sporadic forms of the disease resulting from the rare (perhaps stochastic) conversion of PrPC to PrPSc, or the failure of quality control mechanisms for the suppression or degradation of PrPSc.
Genetic forms (all known examples are associated with mutations in the prion protein gene, PRNP) resulting in a higher probability of conversion to the pathogenic form. Finally, the acquired forms are the result of iatrogenic or oral exposure to PrPSc [6].
Clinical Manifestations
TSEs are progressive neurodegenerative diseases with an incubation period that varies from many months to years. Ninety percent of CJD patients die within a year once clinical signs are diagnosed. Typically, symptoms include progressive dementia, ataxia, and myoclonus. Neuropathologic features include neuronal loss, spongiform degeneration, reactive astrogliosis, and deposition of misfolded PrP species (PrPSc). A key pathologic finding in people with vCJD is extensive lymphoreticular deposition of prion protein that is not seen in other forms of CJD7 and, in particular, this deposit is present during the preclinical phase of the disease [10-12] (Figure 2 and 3).
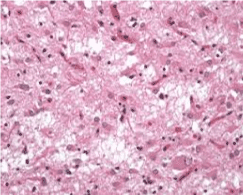
Figure 2. Severe spongiform vacuolation in sporadic CJD results in status spongiosis with extensive neuronal loss, widespread astrogliosis and the resulting collapse of the cerebral cytoarchitecture. (From to: https://www.cjd.ed.ac.uk/sites/default/files/neuropath.pdf)
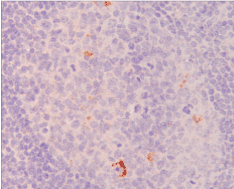
Figure 3. Immunohistochemistry using an antibody to prion protein (brown staining) showing abnormal accumulation in an appendicectomy specimen taken from an individual 8 months before the onset of symptoms of variant Creutzfeldt-Jakob disease [12]
The average age at the onset of sCJD is around 70 years. In addition to the classic symptoms previously described, visual symptoms and pyramidal and extrapyramidal signs can also be part of the clinical manifestations.
In some patients, psychiatric symptoms, behavioral changes, and / or insomnia are also present. The main factors that affect the course of the disease are age of onset, sex, codon 129 of the prion protein gene (PRNP), and the PrPCJD glycotype. The time from diagnosis to death is substantially shorter in patients older than 80 years (median survival 3 months) than in those around 50 years (median survival 7 months). The female population lives about 1 month longer than male patients [4,13-18].
In the case of vCJD, the mean age at onset is 28 years (range 12-74 years), and the median survival after diagnosis is 14 months (range 6-39 months). Onset clinical symptoms include psychiatric, behavioral, and / or sensory symptoms, followed by cognitive decline, cerebellar signs, and involuntary movements as the disease progresses.
Diagnosis
The most challenging aspect of this disease is its diagnosis, the gold standard for definitive diagnosis is histopathological confirmation, but the most recent tests are providing the means for antemortem diagnosis in a less invasive way than brain biopsy.
In 1998, the WHO published diagnostic criteria for CJD, and the diagnosis was based on clinical examination, EEG, and CSF findings. This is somewhat outdated in contemporary medicine because it does not take into account magnetic resonance imaging (MRI) findings, genetic testing, or modern laboratory tests for confirmation of the diagnosis. EEG and CSF protein 14-3 3 tests are included in the criteria, and these studies are commonly performed in the early stages of diagnosis [2].
Periodic acute wave complexes (PSWC) are found in the EEG recordings of approximately two-thirds of patients with sCJD, and therefore have been incorporated into the probable diagnostic criteria for sCJD by the WHO [19].
The sensitivity of the EEG is generally low in sCJD, but when positive it is valuable, giving it a very high specificity [2]. In the study by Heinemann, et al. in Germany it found 37.5% sensitivity and 100% specificity in its cohort of 26 patients. When combined with clinical presentation, examination, and symptoms, the positive predictive value of the diagnosis reached 99% [20].
Initial studies investigating the utility of magnetic resonance imaging (MRI) (Figures 4 and 5) in the diagnosis of CJD used a T2 image pattern consistently. However, technological advance in MRI has made it possible to use FLAIR, diffusion weighted image (DWI) and apparent diffusion coefficient (ADC), improving both the negative and positive predictive value [2,21].
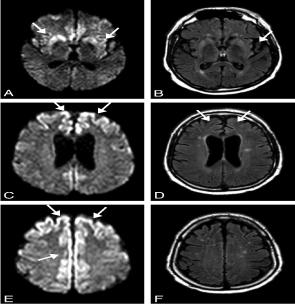
Figure 4. MR imaging scans of a 57-year-old patient with probable sporadic CJD 8 months after onset of disease. On DW MR imaging (A, C, E), there is hyperintensity in the striatum and insula (A), the superior and middle frontal gyri, and the precuneus (C), the superior and middle frontal gyri, the precuneus, and the paracentral lobe (E). On FLAIR (B, D), the insula and cingulate gyrus show increased signal intensity, whereas the hyperintense changes in the precuneus and paracentral lobe, easily identified on DW imaging (E), are questionable on FLAIR (F) [22]
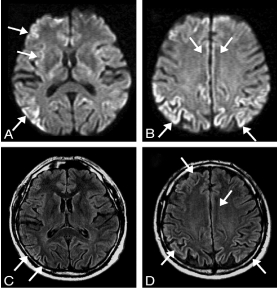
Figure 5. DW (A, B) and FLAIR (C, D) MR imaging of a 63-year-old patient with definite sporadic CJD after 4 months of disease. There is extensive signal hyperintensity, including the cingulate gyrus (A–D), the frontal superior gyrus (A–D), occipital gyrus (A, C), the insula (A, C), the temporal medial gyrus (A, C), and the angular/supramarginal gyrus (B, D). The basal ganglia have normal signal intensity (A, C). The changes in signal intensity are easier to identify on DW (A, B) than on FLAIR imaging (C, D) [22]
MR-DWI is a useful and sensitive test for the diagnosis of sCJD. Hyperintense lesions in MR-DWI in the cerebral cortex and / or basal ganglia (BG) are well recognized findings in patients with sCJD. The presence of hyperintense lesions in MR-DWI BG has been reported to be associated with the clinical course of the disease, giving a shorter duration of the disease and a higher incidence of myoclonus [23-26].
The term "pulvinar sign", or bilateral FLAIR hyperintensity of the pulvinar area, was coined after reviewing 86 patients with vCJD. In that study, 90% of the MRI images were positive for the sign [27]. Although both studies are comparable, MR-DWI is superior to FLAIR in detecting changes in the neocórtex [28].
The diagnostic utility of cerebrospinal fluid biomarkers has been controversial. Studies have shown that amplification methods can detect prions in cerebrospinal fluid, olfactory epithelium, blood, and / or urine in various human prion diseases [29]. In the case of vCJD studies provide a prototype blood test for diagnosis in symptomatic individuals, which could allow the development of large-scale screening tests for asymptomatic prion infection with vCJD [30-32].
The 14-3-3 CSF level is not a useful marker in vCJD. The presence of PrPECJ in the amygdala, detected by immunoblotting and immunohistochemistry, helps distinguish vCJD from other prion diseases [33,34]. Also in one study, the detection of PrPSc in urine specimen was reported only in vCJD patients compared to other forms of prion diseases and other neurological disorders [35]. Carrier status of vCJD has even been reported after the study of human appendages after epizootic bovine spongiform encephalopathy in the United Kingdom [36].
Differential diagnosis
Rapidly progressive dementia (RPD) has a broad differential including vascular, neurodegenerative, autoimmune, infectious, thromboembolic, metastasis/neoplasm, iatrogenic. Vascular conditions like strokes or multiple infarcts or cerebral myeloid angioplasty or hypertensive encephalopathy can lead to rapidly progressive dementia. Vasculitis and intravascular lymphoma can also lead to rapidly progressive dementia. Brain MRI, as well as vascular imaging studies such as MRI angiography and CT angiography, can help diagnose vascular etiologies. Infectious causes like viral encephalitis including herpes simplex virus, HIV dementia, progressive multifocal leukoencephalopathy, fungal infections like central nervous system (CNS) aspergillosis, syphilis, Lyme disease, subacute sclerosing panencephalitis can cause rapidly progressive dementia. Appropriate blood and serological studies can help diagnose infectious causes of RPD. Also neurodegenerative conditions like Creutzfeldt-Jakob disease (iatrogenic, familial), Alzheimer's disease, dementia with Lewy bodies, progressive supranuclear palsy, corticobasal degeneration, neurofilament inclusion body disease, and progressive subcortical gliosis can also cause RPD.
Treatment of prion diseases remains supportive; no specific therapy has been shown to stop the progression of these diseases.
The CJD has a discouraging prognosis, with patients dying within one year of the onset of the disease. CJD should be suspected in any patient with rapidly progressive dementia and myoclonus. However, other diseases that may present with similar clinical conditions such as vascular, infectious, neurodegenerative, neoplastic, autoimmune conditions should be excluded. The MRI findings reported in CJD are abnormal T2 signals in the cerebral cortex and basal ganglia and acutely progressive brain atrophy. Diffusion-weighted imaging is extremely useful in detecting CJD during the very early phase-even before the onset of characteristic clinical findings.
None.
None declared.
We would like to express our sincere gratitude and appreciation to General Hospital of Morelia "Dr. Miguel Silva", for their valuable contribution.
- Spielmeyer W (1992) Die histopathologische Forschung in der Psychiatrie. Klin Wochenschr 1:1817–1819.
- Manix M, Kalakoti P, Henry M, Thakur J, Menger R, et al. (2015) Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus 39: E2. [Crossref]
- Orrú CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, et al. (2014) A Test for Creutzfeldt–Jakob Disease Using Nasal Brushings. N Engl J Med 371: 519-29. [Crossref]
- Zanusso G, Monaco S, Pocchiari M, Caughey B (2016) Advanced tests for early and accurate diagnosis of Creutzfeldt–Jakob disease. Nat Rev Neurol 12: 325. [Crossref]
- Duffy P, Wolf J, Collins G, DeVoe AG, Streeten B, et al. (1974) Possible person-to-person transmission of Creutzfeldt-Jakob disease. N Engl J Med 290: 692–693. [Crossref]
- Head MW (2013) Human prion diseases: Molecular, cellular and population biology. Neuropathology 33: 221–236. [Crossref]
- Du Plessis DG (2008) Prion Protein Disease and Neuropathology of Prion Disease. Neuroimaging Clin N Am 18: 163–182. [Crossref]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, et al. (2008) Physiology of the prion protein. Physiol Rev 88: 673–728. [Crossref]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐β oligomers. Nature 457: 1128–1132. [Crossref]
- Lee J, Hyeon JW, Kim SY, Hwang KJ, Ju YR, et al. (2014) Laboratory Diagnosis and Surveillance of Creutzfeldt–Jakob Disease. J Med Virol 87:175-186. [Crossref]
- Budka H (2003) Neuropathology of prion diseases. Br Med Bull 66: 121–130. [Crossref]
- Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M, et al. (2020) Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis 20: e2-e10. [Crossref]
- Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, et al. (2005) Mortality from Creutzfeldt– Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 64: 1586–1591. [Crossref]
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66: 213–239. [Crossref]
- Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, et al. (1999) Classification of sporadic Creutzfeldt– Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46: 224–233. [Crossref]
- Pocchiari M, Puopolo M, Croes EA, Budka H, Gelpi E, et al. (2004) Predictors of survival in sporadic Creutzfeldt–Jakob disease and other human transmissible spongiform encephalopathies. Brain 127: 2348–2359. [Crossref]
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, et al. (2012) Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 11: 618–628. [Crossref]
- Parchi P, De Boni L, Saverioni D, Cohen ML, Ferrer I, et al. (2012) Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter‐rater study among surveillance centres in Europe and USA. Acta Neuropathol 124: 517–529. [Crossref]
- Wieser HG, Schwarz U, Blättler T, Bernoulli C, Sitzler M, et al. (2004) Serial EEG findings in sporadic and iatrogenic Creutzfeldt-Jakob disease. Clin Neurophysiol 115: 2467-2478. [Crossref]
- Heinemann U, Krasnianski A, Meissner B, Kallenberg K, Kretzschmar HA, et al. (2008) Brain biopsy in patients with suspected Creutzfeldt-Jakob disease. J Neurosurg 109: 735–741. [Crossref]
- Caobelli F, Cobelli M, Pizzocaro C, Pavia M, Magnaldi S, et al. (2015) The role of neuroimaging in evaluating patients affected by Creutzfeldt-Jakob disease: a systematic review of the literature. J Neuroimaging 25: 2–13. [Crossref]
- Tschampa HJ, Kallenberg K, Kretzschmar HA, Meissner B, Knauth M, et al. (2007) Pattern of Cortical Changes in Sporadic Creutzfeldt-Jakob Disease. AJNR Am J Neuroradiol 28: 1114-1118. [Crossref]
- Gao T, Lyu JH, Zhang JT, Lou X, Zhao W, et al. (2015) Diffusion-weighted MRI findings and clinical correlations in sporadic Creutzfeldt–Jakob disease. J Neurol 262:1440-1446. [Crossref]
- Geschwind MD, Potter CA, Sattavat M, Garcia PA, Rosen HJ, et al. (2009) Correlating DWI MRI with pathological and other features of Jakob– Creutzfeldt disease. Alzheimer Dis Assoc Disord 23: 82–87. [Crossref]
- Ukisu R, Kushihashi T, Kitanosono T et al (2005) Serial diffusion-weighted MRI of Creutzfeldt–Jakob disease. AJR Am J Roentgenol 184: 560–566. [Crossref]
- Meissner B, Köhler K, Körtner K, Bartl M, Jastrow U, et al. (2004) Sporadic Creutzfeldt–Jakob disease magnetic resonance imaging and clinical findings. Neurology 63: 450–456. [Crossref]
- Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, et al. (2003) Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol 24: 1560-1569. [Crossref]
- Kallenberg K, Schulz-Schaeffer WJ, Jastrow U, Poser S, Meissner B, et al. (2006) Creutzfeldt-Jakob disease: comparative analysis of MR imaging sequences. AJNR Am J Neuroradiol 27: 1459–1462. [Crossref]
- Kim MO, Geschwind MD (2015) Clinical update of Jakob–Creutzfeldt disease. Curr Opin Neurol 28: 302–310. [Crossref]
- Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, et al. (2011) Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377: 487–493. [Crossref]
- Jackson GS, Burk-Rafel J, Edgeworth JA, Sicilia A, Abdilahi S, et al. (2014) Population Screening for Variant Creutzfeldt-Jakob Disease Using a Novel Blood Test. JAMA Neurol 71: 421-428. [Crossref]
- Brown P, Rohwer RG, Dunstan BC, MacAuley C, Gajdusek DC, et al. (1998) The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 38: 810-816. [Crossref]
- Heath CA, Cooper SA, Murray K, Lowman A, Henry C, et al. (2010) Validation of diagnostic criteria for variant Creutzfeldt–Jakob disease. Ann Neurol 67: 761–770. [Crossref]
- Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, et al. (1999) Investigation of variant Creutzfeldt– Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 353: 183–189. [Crossref]
- Moda F, Gambetti P, Notari S, Concha-Marambio L, Catania M, et al. (2014) Prions in the Urine of Patients with Variant Creutzfeldt–Jakob Disease. N Engl J Med 371: 530-539. [Crossref]
- Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, et al. (2013) Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 347: f5675. [Crossref]