Cardiomyopathies (CM) refer to a group of myocardial diseases associated with significant morbidity and mortality despite marked improvements in medical and device therapies. Efforts to improve therapeutic efficacy and minimize mortality has seen an increase in research for aetiology-specific therapies. A rare but an important aetiology of CM is depositional diseases, which present with a wide spectrum of clinical manifestations depending on the type, location and stage of the disease. It has an unfavourable prognosis and often results into death if left untreated. In the initial stages, it is asymptomatic presenting as diastolic dysfunction and restrictive filling pattern with preserved or mildly depressed systolic function. In the later stages, the disease progresses into the dilated phenotype with bi-ventricular dysfunction. Due to its varied clinical manifestations, diagnosis is often delayed. Except for cardiac amyloidosis, the lack of data on other deposition diseases undermines a holistic understanding of their role in the pathophysiology of CM. Although early diagnosis and targeted treatment may improve patient and clinical outcomes, the lack of clinical guidelines or proven therapies undermine the effectiveness of current clinical management strategies. This literature review and meta-analysis aggregates data on CM due to depositional diseases with a particular focus on pathophysiology, clinical presentation, diagnosis and clinical management. The intention is to improve clinical outcomes and minimize mortality in patients with CM secondary to depositional diseases.
deposition diseases cardiomyopathy, dilated cardiomyopathy, cardiac amyloidosis, hemochromatosis, oxalosis, ochronosis
Deposition diseases refer to a diverse group of disorders resulting from the accumulation of substances in various body organs usually due to abnormal metabolism or degenerative phenomena occurring locally or systematically [1]. They are an uncommon cause of cardiomyopathy (CM) with sporadic reports of myocardial involvement in literature. The initial official recognition of the pathogenic role of deposition diseases in CMs appeared in the 2006 American Heart Association (AHA) scientific statement on contemporary definition and classification of dilated CMs [2]. The paper classified CMs into two categories (primary and secondary) based on the predominant organ involvement. Primary CMs (which include genetic, non-genetic or acquired) solely or predominantly affect the myocardium whereas secondary CMs exhibit pathological myocardial involvement as part of a larger number and variety of a generalized systemic (or multi-organ) disease. Whereas the AHA statement did not explicitly classify deposition diseases as a sub-category of CMs, listed amyloidosis and hemochromatosis among the leading causes of infiltrative and storage CMs respectively, which are sub-categories of secondary CMs [2]. Since then, deposition diseases has attracted a growing research attention as an important aetiology of CMs. However, data on deposition diseases CMs is sporadic, which has undermined a holistic understanding of the disease. However, with the current enhanced clinical awareness and better diagnostic imaging techniques, the incidence of deposition diseases in CM patients is increasing, which underscores the need for an understanding on the disease process as well as improving diagnostic and management strategies [3]. Thus, this review summarizes published evidence on the epidemiology, pathophysiology, clinical presentation, diagnosis and management including a meta-analysis of diagnostic and therapeutic strategies. This review also highlights grey areas of that would benefit from further research to improve the diagnosis and management of CMs secondary to deposition diseases.
Typical characteristic of deposition diseases is the accumulation of endogenous or exogenous substances on various body organs, which if left untreated may potentially lead to organ dysfunction and eventually death. Although numerous deposition diseases exist, few involve the heart and fewer result in CM. The most commonly encountered deposition diseases with pathological myocardial involvement include amyloidosis, hemochromatosis, oxalosis and ochronosis.
Cardiac amyloidosis
Amyloidosis is a degenerative protein-deposition disease in which a specific precursor protein pathologically misfolds from its physiological tertiary structure into a more linear shape dominated by unbranched beta-pleated sheet configuration [4]. These abnormally misfolded proteins aggregate into oligomers and eventually form insoluble amyloid fibrils (serum protein), which accumulate extracellularly in tissues. Both the circulating oligomers (which exert a cytotoxic effect) and the amyloid fibrils (which cause distortion of tissue architecture) lead to tissue dysfunction [4]. Whereas neurologic amyloidoses such as Alzheimer’s and Parkinson’s diseases have received the greatest recognition, there are also a number of systemic amyloidoses affecting virtually all organs except the brain [5,6]. Clinical manifestations vary based on the site of amyloid deposits and the type of precursor protein involved [4].
Many types of precursor protein can affect the heart: (i) light-chain (LC) immunoglobulin; (ii) mutant hereditary transthyretin (TTR); (iii) wild-type TTR; (iv) mutant apolipoprotein; (v) amyloid atrial natriuretic peptide localized to the atrium; (vi) fibrinogen alpha type; and (vii) serum amyloid A (SAA) protein [7,8]. Secondary amyloidosis is typically a consequence of chronic inflammatory conditions such as rheumatoid arthritis, Crohn’s disease or other chronic inflammatory/infectious diseases or familial Mediterranean fever of idiopathic amyloid A amyloidosis [9,10]. Typical organs involved in amyloidosis are liver, kidney, gastrointestinal (GI) tract, nervous tissue and the heart. The presence of amyloid deposits in cardiac tissues varies with the type of amyloid disease (or precursor protein): systemic senile amyloidosis (SSA) and some forms of TTR-related amyloidosis (ATTR) that affect the heart almost invariably while cardiac involvement in amyloidosis light chain (ALC) occurs in about 50% of the cases. In secondary amyloidosis, cardiac involvement is rare, minimal or clinically non-significant [8]. Table 1 presents a list of the most common types of amyloidosis alongside their precursor protein, typical decade of presentation and organs involved.
Table 1. Summary of pathology and presentation of different types of amyloidosis
Amyloid Type |
Precursor of Amyloid Fibril |
Typical Decade of Presentation |
Organs Involved |
AL |
Immunoglobulin light chain |
6th or 7th |
Heart, kidney, liver |
ATTR (V30M) |
Mutant TTR |
3rd or 4th but varies geographically |
Heart (uncommon), peripheral/ autonomic nerve |
ATTR (T60 A) |
Mutant TTR |
6th |
Heart (up to 90%); peripheral/ autonomic nerve |
Wild-Type ATTR |
Wild Type TTR |
7th but may be considered after 5th |
Heart, carpal tunnel syndrome |
ATTR Ile 122 |
Mutant TTR |
6th of older |
Heart, carpal tunnel syndrome |
Apolipoprotein |
Mutant apolipoprotein |
6th or older |
Heart (rare), predominantly Kidney |
Secondary Amyloidosis |
Serum Amyloid A (SAA) |
Any |
Heart (rare), kidney, liver |
Atrial Natriuretic Peptide (ANP) |
ANP |
7th or older |
Heart |
AL: Amyloidosis Light chain; ANP: Atrial Natriuretic Peptide; ATTR: Amyloidosis Transthyretin; TTR: Transthyretin. Tx: Transplantation; Adapted from Banypersad et al. [11].
Types of cardiac amyloidosis
Light chain amyloidosis (AL): Amyloidosis light chain (AL), formally known as primary amyloidosis, is a clonal plasma cell disorder caused by the overproduction and misfolding of antibody light chain fragments. The disease is rare with about 3,000 new cases reported annually in the United States [12]. The median age of diagnosis is 63 years, although the disease can present in patients as early as the third or fourth decade of life [12,13]. The spectrum and pattern of organ involvement is very wide but cardiac involvement occurs in about half of the cases and sometimes it is the only presenting feature [14]. AL may progress rapidly. Low QRS voltages are common, particularly in the limb leads. Typically, thickening of the left ventricular (LV) wall is mild to moderate and rarely greater than 18 mm even in advanced disease. Cardiac AL amyloid deposits leads to a marked elevation of cardiac biomarkers such as B-type natriuretic peptides (BNP) and cardiac troponin, even at the early stage. Cardiac involvement is the most common cause of death in AL and is a major determinant of prognosis. In the absence of cardiac involvement, AL patients have a median survival of about 4 years but prognosis among AL patients with significant elevated BNP and cardiac troponin significantly reduces to about 8 months [15,16]. AL is a more aggressive disease than ATTR, and early diagnosis is crucial because of the high mortality in the absence of early treatment [4].
Transthyretin amyloidosis (ATTR): Hereditary amyloidosis results from mutations in several genes including TTR, fibrinogen, and apolipoprotein although by far the most common cause is variant ATTR due to mutations in the TTR gene causing neuropathy and often with cardiac involvement [11]. ATTR (previously called familial amyloid polyneuropathy) occurs as a result of misfolding of the liver-synthesized precursor protein TTR (previously called prealbumin). ATTR is transmitted as an autosomal dominant trait with high penetrance associated with a late onset CM that is indistinguishable from senile cardiac amyloidosis. More than 100 pathogenic TTR mutations have been described but the most common mutation is the replacement of valine by methionine (Val 122 IIe) [17,18]. Initially reported in Portugal in 1950 [19], ATTR has been described as endemic in Japan and Sweden, and sporadic cases have been reported in many countries [6]. Although the prevalence of ATTR remains unclear, it is almost certainly underdiagnosed because wall thickening may be incorrectly attributed to hypertensive heart disease [11]. Cardiac involvement in ATTR varies by mutation and may be the presenting or the only clinical feature [20].
Wild type ATTR (ATTRwt): Wild Type ATTR (ATTRwt: previously known as systemic senile amyloidosis [SSA], results in the deposition of TTR mainly in cardiac tissues. The main difference between ATTRwt and ATTR is the former is acquired variant while the latter is hereditary mutant variant [4]. The prevalence of the disease is not well defined. The disease typically affects older males and presents as a late onset hypertrophic restrictive CM. Carpal tunnel syndrome, spinal stenosis or both occur in about 50% of ATTRwt patients and often precede clinical presentation of heart failure (HF) by 5 to 10 years but other extra cardiac involvement are very rare [21-24]. ATTRwt has a strong male predominance and the natural history remains poorly understood although there are suggestions of a median survival of about 7 years from presentation [11]. The true incidence of ATTRwt is probably underestimated. However, recent developments in cardiac magnetic resonance imaging (CMRI) have greatly improved detection of cardiac amyloid during life and suggest that ATTRwt is more common than previously thought. ATTRwt accounted for 0.5% of all patients seen at the UK amyloidosis centre until 2001 but now accounts for 7% of 1100 cases with amyloidosis seen since the end of 2009 [11]. It is often an unrecognized cause of diastolic HF in the elderly patients, with up to 25% of patients (≥ 85 years) with ATTRwt showing amyloid deposits on autopsy studies [25]. In a recent study, 39% of hospitalized HF patients (≥ 60 year) with preserved ejection fraction (HFpEF) had grade 2 or 3 uptake on 99mtechnetium-pyrophosphate scintigraphy, which is consistent with ATTRwt [26]. Although echocardiographic findings of ATTRwt maybe indistinguishable from advanced AL, ATTRwt patients have fewer symptoms and better survival [27].
Pathophysiology: Both AL and ATTR-associated cardiac amyloidosis result in diffuse fibril deposition in the heart leading to the thickening of both ventricles [28,29]. The pattern of amyloid deposition in AL is usually sub-endocardial and diffuse while ATTR (especially ATTRwt) can have patchy areas of transmural involvement. Phenotypic expression may vary in particular in ATTR, where a subset may exhibit asymmetric septal hypertrophy mimicking hypertrophic cardiomyopathy (HCM) [30-32]. In cardiac amyloidosis, amyloid deposits occur extracellularly in the interstitium, surrounding the myocytes although there may be depositions in the small intramural coronary arteries. In contrast, in sarcoidosis, amyloid depositions can occur throughout the myocardial tissue. Thus, endomyocardial biopsy is nearly 100% sensitive for the diagnosis of cardiac amyloidosis [33-36].
In some cases, the atria may be involved with inter-atrial septal thickening resulting in poor atrial function and increased rates of atrial fibrillation (AF) as well as the conduction system leading to various degree of heart block and bundle branch block, which manifest more frequently in ATTR than in AL [29,37,38]. The valves usually thicken accompanied by mild to moderate regurgitation. The involvement of pericardial may lead to small pericardial effusions and coronary involvement of the small intramural vessels may lead to ischemia and angina with normal epicardial coronaries that are more common in AL than in ATTR [8,39,40]. Thickened bi-ventricular walls result in a non-dilated ventricle that is stiff and poorly compliant leading to progressive abnormalities in diastolic filling. Systolic dysfunction may be observed in severe and advanced disease. However, LV ejection fraction (LVEF) on echocardiography may be less reliable since reduced end-diastolic volume produces a low stroke volume. For example, LVEF of 50% when starting at a significantly reduced end-diastolic volume (e.g. 70 mL), leads to a significantly reduced stroke volume (35 mL) and thus reduced cardiac output. This may explain why patients with cardiac amyloidosis cannot tolerate reduced heart rates since cardiac output depends on the heart rate [41-43].
Clinical presentation: Irrespective of the precursor protein and disease type, cardiac amyloidosis may present as a restrictive cardiomyopathy (RCM) characterized by progressive diastolic and subsequently systolic biventricular dysfunction and arrhythmia [8]. Typical presentation of cardiac amyloidosis is HF with preserved ejection fraction (HFpEF). Exertional dyspnoea is common although some patients may present with a more right-sided HF symptoms such as lower extremity oedema and ascites. Patients may present with fatigue and weakness associated with low cardiac output and often linked to non-specific symptoms of ageing. Patients with thickened ventricles may be misdiagnosed as having HCM with or without obstruction [41,60]. The initial manifestation of cardiac amyloidosis may be AF (mostly in ATTRwt) or cardioembolic stroke. AF can be present for years before the diagnosis of cardiac amyloidosis is made. Bundle branch block and complete heart block (mostly in ATTR than in AL patients) may require pacemaker implantation. Angina with normal coronary artery can occur while cardiogenic shock due to diffuse ischemia is a rare occurrence [8,39,40]. Elderly patients may present with low-flow, low-gradient aortic stenosis [61].
Extra cardiac involvement such as nephrotic syndrome, autonomic neuropathy, pulmonary or bronchial involvement particularly in AL might cloud cardiac presentation. Pulmonary oedema is uncommon early in the disease process but pleural and pericardial effusion, and atrial arrhythmia are frequent observations. [44-46]. Syncope is a common presentation and a poor marker of prognosis [47]. It is typically exertional or postprandial as part of RCM, sensitive to intravascular fluid depletion from loop diuretics in combination with autonomic neuropathy or conduction tissue involvement (atrioventricular or sinoatrial nodes) or ventricular arrhythmia [48-50]. Disproportionate septal amyloid accumulation mimicking HCM with dynamic LV outflow tract obstruction is rare but well documented [51-54]. Myocardial ischemia may result from amyloid deposits within the microvasculature [55,56]. Atrial thrombus is a common occurrence particularly in AL patients sometimes occurring before AF [57]. Intracardiac thrombus may embolize, causing transient ischemic attacks or strokes and may be an early or even the presenting feature [58]. Anticoagulation is therefore important in the appropriate clinical situation but careful consideration should be given to patients with extensive systemic AL who may have elevated haemorrhaging risk due to factor X deficiency or in some cases with GI involvement [59].
Several other symptoms can help raise clinical suspicion of cardiac amyloidosis (Table 2). Bilateral carpal tunnel syndrome is a common presentation in both AL and ATTR but more so in ATTRwt and can precede clinical HF by several years [21,24,60]. Spinal stenosis is specific to ATTRwt patients due to amyloid infiltration of the ligamentum flavum [22]. In previously hypertensive patients, low to normal blood pressure leading to discontinuation or reduction of anti-hypertensive therapy may suggest possible cardiac amyloidosis. Peripheral and autonomic neuropathy may occur in both AL and ATTR but uncommon in ATTRwt [23]. Other clinical signs and symptoms of AL can include macroglossia and periorbital purpuraor, proteinuria, jaw claudication and GI symptoms of diarrhoea and weight loss [60].
Table 2. Symptoms raising clinical suspicion of amyloidosis cardiomyopathy
Common Clinical Symptoms of Cardiac Amyloidosis |
On echocardiography:
- Low voltage on ECG + thickening of septum/posterior wall > 1.2 cm
- Thickening of RV free wall and valves
|
Intolerance to beta-blockers or ACE-inhibitors |
Low normal BP in patients with a previous history of hypertension |
History of bilateral carpal tunnel syndrome, often requiring surgery |
Light Chain Amyloidosis (AL) |
Transthyretin Amyloidosis (ATTR) |
HF + Nephrotic Syndrome |
White male age ≥ with HFpEF + history of carpal tunnel syndrome and/or spinal stenosis |
Macroglossia and/or periorbital purpura |
African American age ≥ 60 with HFpEF without a history of hypertension |
Orthostatic hypotension |
New diagnosis of hypertrophic cardiomyopathy in elderly patient |
Peripheral neuropathy |
New diagnosis of low flow, low gradient aortic stenosis in elderly patient |
Monoclonal gammopathy of undetermined significance |
Family history of ATTR amyloidosis |
ACE: Angiotensin-Converting Enzymes; AL: Light Chain Amyloidosis: ATTR: Transthyretin Amyloidosis; BP: Blood Pressure; ECG: Electrocardiogram; HF: Heart Failure; HFpEF: Heart Failure with Preserved Ejection Fraction; RV: Right Ventricular
Clinical evaluation: Clinical evaluation of cardiac amyloidosis involves three features: (i) demonstration of amyloid protein in tissue specimen; (ii) identification of affected organs; and (iii) definition and type of amyloidosis [3]. According to the AHA 2016, cardiac amyloidosis is clinically suspected on echocardiography, usually performed because of the presence of HF symptoms or for screening purposes when the diagnosis of amyloidosis has been established in extra cardiac organs. Typical echocardiography features of cardiac amyloidosis include thickened bi-ventricular walls in the setting of normal ventricular size, bi-atrial dilation, the presence of pericardial effusion, and valvular thickening without significant dysfunction. Increased echogenicity of the myocardium is non-sensitive when considered in isolation but should raise clinical awareness in the presence of typical findings listed previously although it has become less discernible in newer echocardiographic modalities. Thickened ventricular walls in the absence of known causes such as hypertension or aortic stenosis raises the clinical suspicion for an infiltrative disease process. Doppler studies demonstrate impaired LV relaxation and restrictive filling pattern. Initially, LV systolic function may be preserved but it gradually declines as the disease progresses [11,62,63]. On ECG, low QRS voltage amplitude occurs in approximately 50% of patients with cardiac amyloidosis despite the presence of thickened ventricular wall on cardiac imaging. Other common ECG abnormalities include a pseudoinfarct pattern in the precordial leads, AF, and atrioventricular (AV) conduction abnormalities [3].
Nuclear imaging with technetium pyrophosphate has been shown to be insensitive. However, if uptake is present, amyloid infiltration should be considered, depending on the clinical setting, and may be useful in distinguishing between AL and ATTR [64,65]. Cardiac MRI shows a characteristic late gadolinium enhancement (LGE) pattern that is diffuse and sub-endocardial that does not follow any particular coronary distribution. LGE can also be seen in the RV and the atrial walls, and can be transmural and patchy in ATTRwt [4]. However, the accuracy and utility of cardiac MRI in the diagnosis of cardiac amyloidosis remains uncertain although it could help identify the extent of cardiac involvement in patients with an established diagnosis of amyloidosis [66,67]. BNP levels may also be helpful in the diagnosis and follow-up of cardiac amyloidosis patients. Elevated BNP levels have been observed even in the absence of clinical evidence of clinical HF or increased wall stress suggesting it might be caused by elevated ventricular filling pressure and by direct myocyte damage due to extracellular amyloid deposits. Elevated BNP suggest cardiac involvement with sensitivity and specificity of 93% and 90% respectively [68,69]. Elevated BNP may also be helpful to predict the development of clinical cardiac involvement in the future [70]. BNP levels decrease with chemotherapy of AL possibly suggesting organ response [71].
Diagnostic gold standard for cardiac amyloidosis is histologically from endomyocardial biopsies (EMB) of abdominal fat pad, gingiva, rectum, bone marrow or other affected organs such as heart, liver and kidney. However, its main limitations are it has 1% risk of ventricular perforation leading to cardiac tamponade and it is usually unavailable in many centres [4]. Other definitive diagnosis for cardiac amyloidosis include light microscopy showing amorphous pink deposits in the interstitium. With Congo red staining, amyloid fibrils produce apple-green birefringence under polarized microscopy. Other confirmative test is electron microscopy and proteomic typing by mass spectrometry [72,73]. EMB that identifies amyloid protein provides the definitive diagnosis of cardiac amyloidosis. In patients with extra-cardiac tissue-proven systemic amyloidosis, echocardiographic or cardiac MRI findings suggestive or infiltrative CM (such as wall thickness > 12 mm) can support the diagnosis of cardiac amyloidosis obviating the need for EMB [74].
After establishing the diagnosis of cardiac amyloidosis, the next diagnostic step is to identify the type of amyloidosis, which is instrumental in determining both treatment strategy and prognostication. Immunohistochemistry can be performed on tissue samples with antibodies against amyloid A, light chains and TTR amyloid [62]. If TTR is detected, DNA mutational analysis can help distinguish between senile and hereditary amyloidosis [73]. The presence of serum or urine monoclonal gammopathy suggests the presence of AK amyloidosis but does not establish the diagnosis. In reactive (AA) amyloidosis, deposited protein is serum amyloid protein A, an acute phase protein that is normally soluble and whose plasma concentration is the highest during inflammation. AA amyloidosis is a complication of a number of inflammatory diseases and infections including tuberculosis, chronic osteomyelitis and autoimmune diseases. AA amyloid deposits are primarily in the liver, spleen and kidney and rarely affect the heart [3].
Clinical management: Generally, cardiac amyloidosis has a poor prognosis, which differs based on amyloid type, and availability and response to therapy. Treatment for the most part is supportive therapy – modified HF treatment including device therapy. Other treatment options include therapies that suppress the production of amyloid fibril precursor protein such as chemotherapy in AL amyloidosis, and novel strategies to inhibit amyloid fibril formation or directly targeting amyloid deposits or stabilize the precursor protein especially ATTR. Cardiac transplantation, although not in common use, can very successful in carefully selected patients [3,11].
Supportive treatment includes standard HF therapy. This may be of limited benefit or even detrimental in cardiac amyloidosis, which requires careful monitoring to avoid significant drug interactions and adverse side effects [76]. The evidence for the use of angiotensin-converting enzyme inhibitors (ACE-I), angiotensin receptor blockers (ARBs) or beta-blockers. Toleration for HF medication may be poor and may aggravate postural hypotension or renal function. Some patients may develop RCM leading to heart rate dependent cardia output and these patients may find it difficult tolerating beta-blockers. Digitalis and calcium channel blockers may be selectively concentrated in amyloidotic tissue and may be relatively contraindicated because of increased toxicity [76-78]. The maintenance of adequate filing pressure is crucial because of restrictive physiology, balancing peripheral oedema and renal impairment with salt/water restriction and judicious use of diuretics. Patient education and participation with conjunction with HF team is critical to successful manager. Maintenance of adequate BP with an alpha-antagonist (such as midodrine) may permit higher doses of loop diuretics, especially in patients with autonomic neuropathy [79]. Device therapies such as pacemakers or implantable cardioverter defibrillators (ICD) may not prevent sudden cardiac death due to electromechanical dissociation [80]. In the absence of evidence, pacing indications remain within the current standard guidelines. Bi-ventricular pacing appear to have little role but although may be the ideal pacing option to avoid decompensation of the stiffened ventricles due to induced dyssynchrony from RV pacing [81].
Amyloid specific treatment includes reducing the production of amyloid fibril precursor-protein or inhibition of amyloid formation [11]. In AL patients, treatment for reducing the supply of amyloid-fibril precursor protein targets the clonal plasma cells using cyclical combination chemotherapy or high dose therapy with autologous stem cell transplantation. Most chemotherapy regimens consist of dexamethasone together with alkylator. However, the use of dexamethasone should be cautious because of the risk of fluid overload in the absence of adequate and rapid changes to diuretic therapy requiring close coordination between teams treating haematology and cardiology to improve outcomes [82]. High-dose melphalan followed by autologous stem-cell transplantation is generally contraindicated in patients with advanced cardiac amyloidosis although autologous stem cell transplantation may be the best treatment for suitable patients [83-85].
Besides AL, AA amyloidosis is the only other type in which available therapies can effectively suppress the production of fibril precursor. Anti-inflammatory therapies such as anti-tumour necrosis factor agents in rheumatoid arthritis can significantly suppress the production of serum amyloid A protein although with limited clarity on the effect on cardiac involvement, which is very rare in AA amyloidosis. Since the liver almost exclusively produces TTR, ATTR has become a focus of novel drug developments to suppress TTR production through silencing RNA and antisense oligonucleotide therapies [86]. Finally, the concept of treatment by inhibiting amyloid formation is based on the observations that amyloid fibril formation involves conformational transformation of precursor proteins into a completely different form with predominant beta-sheet structure. This conversion may be inhibited by stabilizing the fibril precursor through specific binding to a pharmaceutical has been explored in ATTR with promising results [87,88]. Finally, following treatment that prevents production of new amyloid, treatment that targets amyloid deposits by immunotherapy may mobilize AL deposits in a majority of patients. However, cardiac clearance of amyloid is slow and echocardiographic evidence of improvement has not been confirmed. The concept of passive immunotherapy to enhance clearance has proved successful in experimental models and is currently in clinical development [11].
Haemochromatosis: Haemochromatosis is an autosomal recessive disorder associated with abnormal deposition of iron in parenchymal organs causing organ toxicity and dysfunction or primary iron-overload CM [89,90]. The disease is almost exclusive to populations of Northern Europe descent [91]. Primary iron-overload CM was first described in the 18th Century characterized by a clinical triad of cirrhosis, hyperpigmentation of the skin and diabetes presented in about 15% of the patients [90]. In the 19th Century it was recognized that haemochromatosis is hereditary in nature and occurs due to mutations in the genes encoding proteins involved in iron metabolism [92,93].
Types of Haemochromatosis
Haemochromatosis has four sub-types caused by increased GI absorption of iron into the bloodstream and reduced activity or synthesis of hepcidin protein that regulates the entry of iron into circulation [94,95]. Type 1 is due to mutations of the HFE gene on chromosome 6 that accounts for more than 80% of cases of haemochromatosis and corresponds to classical hereditary haemochromatosis. Type 2: is due to mutations of the HJV gene on chromosome 1 that encodes hemojuvelin (subtype 1A) or in the HAMP gene on chromosome 19 that encodes hepcidin (subtype 2B). Type 3 is due to mutations of the TfR2 gene on chromosome 7 encoding transferrin receptor 2. Finally, Type 4 is due to mutations of the SLC40A1 gene on chromosome 2 that encodes ferroportin. All these mutations are inherited in an autosomal recessive pattern with the exception of Type 4, which is inherited in an autosomal dominant pattern [94]. Types 1, 3 and 4 present in adulthood and usually during the fourth or fifth decade of life while Type 2 (also known as juvenile haemochromatosis) presents much earlier in the second or third decades of life and its phenotype is much more severe [93]. The classical triad of haemochromatosis (cirrhosis, bronze skin and diabetes mellitus) but it is extremely variable and depends on several interfering genetic and non-genetic factors especially in Type 1. In Types 1 and 3, hepatic involvement predominated while in Type 2, endocrine and cardiac complications predominate and HF is a frequent cause of death before the third decade of live [93].
Pathophysiology: In haemochromatosis, inappropriately high duodenal iron absorption compared with the total body iron content leads to iron overload [96]. During iron overload, transferrin (iron-binding blood plasma glycoproteins that controls the level of iron in biological fluids), which is normally about 30% saturated, becomes fully saturated leading to the appearance of the toxic non-transferrin-bound iron species in the circulation [97]. The negative feedback mechanisms that regulates transferrin bound iron uptake does not control cellular uptake of non-transferrin-bound iron together with the lack of an iron excretory mechanism leads to intracellular iron accumulation. The uptake of iron from non-transferrin-bound iron species in hepatocytes, cardiomyocytes and endocrine gland cells results in tissue iron accumulation and ultimately the deleterious effects of iron overload [98]. During iron overload, iron in the form of ferrous iron (Fe2+) diffuses into the cardiomyocytes via the voltage-dependent L-type calcium channels [99]. Myocardial iron overload occurs later than hepatic iron overload due to slower myocardial iron uptake. Initially, iron accumulates in the ventricular myocardium then in the atria myocardium as well as affects the conducting system but to a lesser extent relative to the myocardium [97]. The epicardium has a higher iron concentration than the sub-endocardium but the difference becomes blurred in severe iron overload CM [100]. Pathological iron deposition begins initially within the epicardium then extends to the myocardium and then endocardium, which explains the preservation of systolic function until very late in the disease process [95].
The cardiomyocytes stores iron in the form of ferritin (the primary site of iron storage), hemosiderin and labile cellular (free) iron. Labile iron leads to the formation of reactive oxygen species (ROS) through the Fenton reaction that converts ferrous to ferric iron with the generation of the toxic hydroxyl radical [94]. Once the cellular antioxidant capacity is exceeded, the rapid Fenton reaction catalyses iron to produce hydroxyl iron (an extremely free radical species), which causes lipid peroxidation producing membrane permeability alterations. These modifications create a leak of hydrolytic enzymes that initiate dell damage and subsequently cardiomyocyte death. In the presence of myocardial ischemia, iron overload can accelerate ischemia-induced reperfusion injury and may lead to an autocatalytic process resulting in a cardiomyopathic process [101,102]. In addition, increased ferrous iron transportation via the L-type calcium channels may cause derangement of cardiomyocyte calcium transportation and impaired excitation-contraction coupling, which in turn may contribute to the development of diastolic and systolic ventricular dysfunction association with iron-overload CM [103]. The end-result of the previously described processes is the development of CM characterized mainly by LV dysfunction [104,105]. In addition to direct myocardial injury, iron overload may exert an indirect effect on the myocardium via its effect on other organs. Hepatic dysfunction, endocrinopathies (diabetes mellitus, hypo- and hyper-thyroidism, hypo-parathyroidism), and immune deficiency all due to iron overload contribute to the pathophysiology of iron overload CM [106,107]. Besides iron overload, other mechanisms such as immune-inflammatory and genetic factors appear to interfere in the pathogenesis of iron overload C< such as myocarditis, the HLA genotype and the apolipoprotein E genotype [100,105,108].
Iron overload CM presents in two phenotypes, dilated and restrictive. The dilated phenotype results from a process of LV remodelling leading to chamber dilatation and depressed LVEF while the restrictive phenotype results from diastolic dysfunction with restrictive filling, preserved LVEF, pulmonary hypertension and subsequent RV dilatation [104]. In addition to these typical manifestations, two phenotypes may be followed by several other manifestation including conduction system abnormalities, tachyarrhythmias and perimyocarditis [109-111]. In the early stages of iron overload CM, myocardial iron overload manifests as diastolic LV dysfunction with elevated LV filling pressures [112,113]. If the cause of iron overload persists with no appropriate treatment, a majority of patients go on to develop LV remodelling ultimately leading to LV dilatation and depressed LVEF (the dilated phenotype) [104,114]. In fewer than 10% of older patients with severe iron overload, restrictive LV dysfunction leads to pulmonary hypertension, RV dilatation and right-sided HF without LV anatomic remodelling with preserved LVEF (restrictive phenotype) [114,115]. The development of dilated or restrictive phenotype depends on the interaction between the main disease and additional immuno-inflammatory and molecular factors such as myocarditis (dilated phenotype) [94].
Clinical presentation: Patients with iron-overload CM often are asymptomatic in the early stages of the disease. Excessive or chronic iron overload may result into irreversible HF if not detected early; thus, early identification is very important to improve the efficacy of clinical management [89]. Once HF develops, there is a marked deterioration. Cardiac haemochromatosis may result into DCM with dilated ventricles, low LVEF and low fractional shortening [116,117]. Initially, patients may experience exertional dyspnoea due to LV diastolic dysfunction with restrictive hemodynamics and increased filling pressures (restrictive phenotype). The dilated phenotype with depressed LVEF develops as the disease progresses. Bi-ventricular failure may ensue causing pulmonary congestion, peripheral oedema and hepatic congestion [118]. Pericardial constriction or tamponade due to pericardial iron accumulation may lead to rapid clinical deterioration [119]. Angina pectoris in the absence of coronary artery disease responding to venesection may also manifest [120]. Excessive deposition of iron in the entire cardiac conduction system, particularly the AV node, may occur. Complete AV block due to iron deposition may require permanent pacemaker therapy [121]. Iron deposition in cardiac tissues may also lead to non-homogenous electrical conduction and repolarization accompanied with atrial and ventricular tachyarrhythmias [122]. Persistent iron overload causes a reduction in calcium-dependent L-type Ca2+ current causing bradycardia, altered electrical conduction and AF [123]. Paroxysmal AF is the most common arrhythmia seen in patients with cardiac haemochromatosis. The prevalence of ventricular arrhythmias correlates with LV dilatation and depressed LVEF. Sudden cardiac death may develop in some patients [124]. However, there is no association between cardiac haemochromatosis and ischemic heart disease or myocardial dysfunction [125,126].
Clinical evaluation: A high index of suspicion is necessary to identify and categorize iron overload CM secondary to haemochromatosis. Diagnosis can be very challenging in the early stages of the disease. Accurate assessment of organ specific iron overload will be helpful [95]. According to AHA diagnostic statement on DCM, upon clinical suspicion, the diagnosis of haemochromatosis associated iron overload CM begins with biochemical markers (serological testing) and includes investigation of end-organ involvement, followed by transthoracic echocardiography with complete LV diastolic function assessment including tissue velocity measurements of the mitral annulus, and finally cardiac MRI to confirm or exclude myocardial iron overload [3].
Serum transferrin saturation – serum iron/total iron bonding capacity of >45% and elevated serum ferritin > 200 µg/L in men or 150 µg/L in women supports diagnosis according to the AHA guidelines for specific forms of DCM [3] and the American College of Physicians (ACP) guidelines for hereditary hemochromatosis [127,128] guidelines. However, serum iron studies poorly correlate with disease severity and uncertain in some patient populations. Transferrin saturation can miss a significant population of patients homozygous for HFE mutations [129]. It may also be elevated along with ferritin levels in Asian and pacific Islanders without HFE mutations and thus have uncertain significance in these populations [130]. Upon suspicion for hereditary haemochromatosis especially with a known family history of haemochromatosis, testing for the haemochromatosis genotype should be performed [3].
Diagnosis of cardiac dysfunction may be possible with echocardiography. As iron overload proceeds, echocardiography may show biventricular dilatation and progressive evidence of RCM from myocardial damage [131]. Echocardiographic evidence of ventricular diastolic dysfunction can detected early before systolic dysfunction by tissue Doppler signals [132,133]. Diagnosis of cardiac iron overload is possible with cardiac MR (the only non-invasive modality that can assess quantitatively myocardial iron load) in which decreased T2 signal correlates with myocardial iron-infiltration and depressed LV function. Cardiac MRI is useful in identifying when therapy is indicated and serial imaging can measure response to medical therapy [3]. The traditional gold standard for making the diagnosis of liver iron overload is tissue biopsy but iron deposit in the heart can be patchy [134]. Thus, tissue biopsy may miss the areas of deposition and provide a false negative result.
Clinical management: Generally, the treatment for iron overload states targets the prevention or reversal of cardiac dysfunction [136-139]. Removal of excess iron from the tissues in these patients minimizes the generation of free radicals to minimize organ damage [140,141]. The established treatment for haemochromatosis is phlebotomy and chelation therapy for secondary iron overload. Clinical therapy recommendations are well outlined in the American Association for the study of liver diseases haemochromatosis practice guidelines [142]. Therapeutic phlebotomy is started with serum ferritin levels ≥ 300 µg/l in men and ≥ 200 µg/l in women irrespective of the presence or absence of symptoms [139]. Therapeutic phlebotomy consists of removing one (1) unit of blood (450-500 ml) weekly until the serum ferritin level is 10 to 30 µg/l and maintenance of serum ferritin level ≤ 50 µg/l after periodic blood removal [139]. Each unit of blood removed depletes 200 to 250 mg of iron from the blood, which then mobilizes an equal amount of iron stored in the tissues to form haemoglobin [143]. However, patients with ferroportin mutation associated iron overload may not tolerate a more aggressive therapy [144].
In addition to phlebotomy, dietary management should be included in the therapy. Avoidance of medicinal iron and iron supplements [139]. Eating large amounts of vitamin C rapidly mobilizes iron from the heart, increases free radicals production and causes life-threatening cardiac arrhythmias [145,146]. Thus, supplemental vitamin C should avoided although vegetables and fruits rich in vitamin C may be ingested [147]. Alcohol increases iron absorption and some red wines contain high iron content [148,149] and both should be avoided. Seafood such as uncooked shellfish should be avoided [149]. Patients with CM and HF should be treated with a low sodium diet [150]. Haemochromatosis patients with HF should be managed with standard medical therapy for HF as per the current HF guidelines [150]. Cardiac transplantation is a therapeutic option for haemochromatosis patients with HF refractory to optimal medical therapy and cardiac resynchronization therapy [150-152].
The initial description of the deposition of calcium oxalate crystals in human tissues was made in 1925 in the kidney of a 4.5 years old child who had multiple urethral, ureteric and bilateral renal calculi [153]. Chou and Donohue [154] coined the term “oxalosis” in 1952 to describe the clinical condition in which calcium oxalate crystals deposits in the kidney, and later Scowen et al. [155] used the term for cases with disseminated extra-renal calcium oxalate deposits. The present definition of oxalosis is a rare metabolic disorder characterized by the inability of kidneys to eliminate calcium oxalate crystals through the urine or enhanced production of oxalic acid leading to hyperoxalemia and a deposition of calcium oxalate in different body organs [156,157]. Oxalate is a by-product of normal metabolism. It is produced endogenously as an end-product of the metabolism of amino acids and absorbed by the stomach, small bowel and colon from dietary sources [158]. In healthy individuals, up to a half of urinary oxalate is derived from dietary sources. Oxalate can be excreted dissolved in urine, precipitated with calcium in the stool or metabolized by gut microbiota [158]. Under normal conditions, all oxalate absorbed from dietary sources is excreted through the urine. Normal urinary oxalate excretion is variable but a value of 40-45 mg/day (0.45 mmol)) is considered hyperoxaluria [159]. Calcium oxalate deposits on the myocardium or the conduction system can potentially lead to life-threatening arrhythmias, CM and HF. The frequency and severity of cardiac oxalosis remain poorly described with the lack of organized data beyond sporadic literature reports. Current information is limited case reports and small case series, which are restricted to CM, valvular diseases and conduction abnormalities [161].
Types of Oxalosis: Oxalosis can be classified into two types: (i) primary oxalosis; and (ii) secondary oxalosis. Primary oxalosis results from hyperoxaluria, described as an inborn error of metabolism transmitted in an autosomal recessive fashion in which the liver cannot create enzyme peroxisomal alanine-glyoxylate aminotransferase or the enzyme does not work properly [160,161]. The enzyme is responsible for regulating the production of oxalate. Its deficiency leads to an increase in the production of oxalate causing excessive amounts to be excreted in urine causing recurrent urolithiasis and nephrocalcinosis. The urinary daily excretion of calcium oxalate is raised constantly above the maximum normal of 40 mg [162]. In the urine, oxalate combines with calcium to form insoluble calcium oxalate crystals (kidney stones) that often damage the kidneys resulting in kidney failure. As progressive renal insufficiency occurs, reduced urinary oxalate excretion in turn results in marked increases in plasma oxalate concentrations and the consequence of systemic calcium oxalate deposition mainly in the muscoskeletal, cardiovascular and peripheral nervous systems, causing systemic oxalosis [157,161]. If left untreated, oxalis can potentially lead to death [160,161].
Secondary (also enteric) oxalosis on the other hand results from several factors including the interaction with environmental factors, underlying primary diseases or organ failure in patients with no personal or family history of oxalosis. The main secondary causes include: (i) dietary hyperoxaluria; (ii) enteric hyperoxaluria; or (iii) kidney failure. Dietary hyperoxaluria usually is the consequence of the consumption of foods high in oxalate content (ascorbic acid [vitamin C], ethylene glycol, xylitol and methoxyflurane), which can increase the levels of oxalate in urine and increase the risk of the formation of calcium oxalate crystals [163]. Enteric hyperoxaluria occurs as a result of certain intestinal diseases such as Crohn’s disease and Short Bowel Syndrome that can cause an increase in the absorption of oxalate from foods, thereby increasing the amounts of oxalate excreted in the urine [159]. Finally, kidney failure due to reasons other than hyperoxaluria in which oxalate can no longer be excreted from the body, can lead to oxalosis [159,161].
Pathological features: The primary route of oxalate elimination is via glomerular filtration. Thus, kidney dysfunction is the most common manifestation of oxalosis. Although cardiac involvement is uncommon, it is a major source of mortality in patients. Data on oxalosis with cardiac involvement is lacking and sporadic reports in literature are insufficient to paint an exact picture of the pathogenic mechanisms. In fact, recent guidelines on myocardial disease from both the AHA and ESC do not include calcium oxalate as a cause of CM [164,165]. Despite the lack of data, studies on patients with hyperoxaluria may provide insightful account of cardiac abnormalities in patients with oxalosis. In particular, a recent retrospective review of an institutional database for 103 patients with hyperoxaluria between 1948 and 2006 revealed several important cardiac abnormalities associated with calcium oxalate deposits on the myocardium [161].
In patients with oxalosis, critical plasma concentration for calcium oxalate of 30-45/µmol/L is easily exceeded as a result of compromised renal function and subsequent extra-renal oxalate deposition [166-168]. Increased calcium oxalate deposits on cardiac tissues has been associated with increased LV mass index (LVMI), impaired LV and RV function, diastolic function, left atrium enlargement, increased wall thickness suggesting myocardial infiltration and rhythm abnormalities. Hormonal and metabolic abnormalities associated with end-stage renal disease may lead to pulmonary vascular abnormalities resulting in pulmonary hypertension. Numerous case reports and limited case series describe calcium oxalate deposits on the myocardium lead to increased wall thickness, restrictive CM, HF, tricuspid and mitral regurgitation, conduction defects ventricular tachycardia, impaired bi-ventricular function [160,162,169-176]. Co-morbid renal failure and hypertension in oxalosis patients may also affect cardiac thickness and diastolic function. Cardiac histopathology analyses describe calcium oxalate deposition within the conduction system, the small intra-myocardial vessels and myocardium [157]. Conduction system abnormalities tachyarrhythmias, bradyarrhythmia, and CM. Secondary inflammatory response to oxalate deposits may also induce thickening and degeneration of collagen fibres, fatty infiltration, mononuclear cell infiltration and frank fibrosis [157,177].
For a better understanding of the pathophysiology of oxalosis CM, there is need for further prospective trials enrolling a large sample of oxalosis to determine or clarify the following concerns. (i) The time between diagnosis of oxalosis and the onset of cardiac manifestations. (ii) Plasma cut-off values for cardiac oxalate deposition associated with cardiac manifestations. (iii) Echocardiographic modalities that have greater sensitivity and specificity in diagnosis of cardiac abnormalities in oxalosis patients such as vector velocity imaging and strain of the LV and RV. (iii) Diagnostic role of cardiac computed tomography or cardiac MRI in providing additional information and evidence of vascular involvement that should obviate the need for myocardial biopsy, the current diagnostic gold standard. (iv) Dose-response of calcium oxalate deposition with higher plasma levels of oxalate causing higher levels of cardiac dysfunction, and whether, liver and kidney transplant may result into the regression of oxalate deposition, which is consistent with other deposition diseases such as haemochromatosis and cardiac amyloidosis.
Clinical evaluation: Oxalosis can present with a multitude of symptoms depending on the location of oxalate deposition, severity and progression of the condition. Clinical presentation of oxalosis patients with cardiac involvement varies widely depending on the type and stage of the disease. They may present with atypical chest pain, exertional dyspnoea, a history of hypertension, elevated plasma oxalate (43.4 µmol/L), decreased renal function with increased serum creatinine (3.39 mg/dl), and reduced glomerular filtration rate (72.3 ml/min) but with a normal mean blood pressure [161]. There are no expert consensus guidelines for the diagnosis and management of oxalosis CM. However, limited data based on findings of case reports and case series reveal that diagnosis may include a combination of tests such as serum tests for oxalate concentration, ECG and non-invasive imaging. ECG may demonstrate sinus tachycardia with atrioventricular sequential pacing [160], AV block progressing into a complete heart block [162]. Transthoracic echocardiography reveals mildly dilated LV with moderate severe systolic function, marked concentric thickening of both the LV and RV with patchy echodense speckled reflections in the myocardium, moderate pericardial effusion with no signs of cardiac tamponade, moderate tricuspid regurgitation [160]. Coronary angiography reveals high-grade stenosis but uncommon while chest x-ray reveals increased cardiothoracic ratio, pulmonary venous congestion (or hypertension) and bilateral pleural effusion [160,161]. These findings are sporadic based on case reports with no randomized controlled trials to confirm these diagnostic features.
Clinical management: Clinical management of oxalosis CM lacks established guidelines and well-defined treatment strategies. Current treatment strategies based on outcomes from case reports suggest treatment with vitamins and minerals can help to reduce or prevent oxalate accumulation in the body. Vitamin B6 can reduce oxalate excreted in the urine and oral preparations of phosphates and citrate can be useful in preventing oxalate crystals from developing. Other medications such as thiazide diuretics may be necessary contingent upon abnormalities in the urine [177]. For oxalosis CM due to primary hyperoxaluria, prescription doses of pyridoxine have been shown to decrease urine oxalate levels by approximately 30% [178]. Urine oxalate levels in individuals on pyridoxine should be monitored monthly to assess individual patient’s responsiveness to this drug. Symptoms related to oxalosis-induced oxalate arthritis have been described to be alleviated via the use of NSAIDs, colchicine and steroids.
Alkalization of urine with alkali citrate can reduce urinary calcium oxalate saturation by forming complexes with calcium thus reducing calcium oxalate crystals or nephrocalcinosis [178]. Dietary changes (limiting or eliminating foods high in oxalate from the diet) may be effective in reducing the amount of oxalate excreted in urine in patients in whom accumulation of oxalate is due to dietary causes [177]. If kidney function is not impaired, increasing fluid intake (>3 L/m2 per day distributed throughout the day) will help to flush the kidneys and prevent oxalate build up and the formation of calcium oxalate crystals [178]. Dialysis is a temporary procedure used to compensate for kidney failure by imitating kidney function by removing extra fluids and toxins from the blood but cannot remove oxalate that accumulates elsewhere in the body [156,177]. For patients with kidney failure, a kidney and liver transplant may be necessary although there are cases of persistence echocardiographic abnormalities 12 months after transplant [156].
Prevalence: Ochronosis (also alkaptonuric) is a very rare autosomal metabolic disorder caused by the deficiency of homogentisic acid (HGA) oxidase (alkaptonuria) affecting between 1 in 250,000 and 1 in 1 million births [179]. The highest incidence occurs in Slovakia and the Dominican Republic, where the incidence reached 1 in 19,000 births. It is a consequence of a discrete mutation in chromosome 3q21-3q23, leading to a deficiency in the homogentisate 1, 2 dioxygenase (HGO) [180]. The deficiency of the enzyme (HGO) leads to an increase in the levels of HGA (a by-product of tyrosine and phenylalanine metabolism), accumulates and become polymerized and be systematically deposited within various tissues of the body (ochronosis) [181]. In the urine, the HGA is oxidized to a melanin-like material causing the urine to acquire a blackened appearance when exposed to air or reducing agents. In the body, the HGA undergoes polymerization causing it to accumulate in connective tissues, which appear ochre-coloured (yellow/brown) material under microscope [182,183]. As the disease progresses, tissue deposition of polymerized HGA eventually will lead to the progressive degeneration of all affected body systems. Although the term ochronosis has been applied to alkaptonuria, it also refers more generally to any deposition of an ochre-coloured material in connective tissues [181,182]. Excess polymerized HGA deposits within various cells types including endothelial cells, macrophages, fibroblasts and smooth muscle cells. In patients with alkaptonuria, pigment occurs in heart valves, healed myocardial infarcts, atherosclerotic vascular lesion, and throughout non-atherosclerotic arterial walls. [182-186].
Pathophysiology: Cardiac manifestations is very rare with ochronosis described in heart valves, aorta, pericardium, endocardium and coronary arteries [180,187,188]. Two frequently described pathological features associated with ochronotic cardiac involvement are aortic stenosis and coronary artery calcification. Aortic stenosis is the most common cardiac manifestation with a number of case reports describing an increase in the prevalence of aortic stenosis in comparison to the general population [180]. In the Cardiovascular Health Study enrolling 5,621 patients (≥ 65 years), 1.8% had aortic stenosis [189] while in a series of 76 patients with alkaptonuria, 25% of patients (≥ 65 years) had aortic stenosis [190]. In a recent review of 175 cases of alkaptonuria, 35% had aortic valve disease, 19% aortic stenosis, 11% aortic regurgitation and 5% aortic valve disease or aortic valve replacement for unspecified reasons [180].
The pathogenesis of cardiac manifestations in ochronotic patients is unclear, although several theories have been proposed. It is hypothesized that ochronotic pigment is initially deposited in fibrocytes, macrophages, smooth muscle cells and the extracellular matrix. These pigment-laden cells degenerate to release the pigment extracellularly acting as a chemical irritant to produce a pro-inflammatory reaction or as a direct enzyme inhibitor that causes alteration in cartilage metabolism resulting in dystrophic calcification and fibrosis of the cardiac valve leaflets [191]. The predominant presence of ochronotic deposition in areas of turbulent flows where there are eddy current such as in sinotubular junction, which plays a role in diastolic coronary filling. This may explain the deposition of ochronotic pigment in the ostia of the coronary arteries and aortic valve leaflets whereas there is minimal deposition in venous circulation [180]. Thus, vascular flow dynamics dictate the site of pigment deposition resulting in subsequent microvascular injury [192].
Clinical evaluation: Diagnosis for ochronotic CM lacks well-defined guidelines. In literature, only a handful of case reports have described diagnosis of cardiac involvement in ochronotic patients. The classic clinical triad of ochronosis is (i) homogentisic aciduria (urine blackens on standing when oxidized or alkalinized); (ii) eumelanin-like pigmentation of skin, sclera, cartilages; and (iii) degenerative ochronic arthropathies usually manifesting in the fourth decade of life due to decline in renal clearance with age [185]. There is a wide clinical variable among patients that may be explained by 84 different mutations identified in the HGO gene. Patients also exhibit different rate of renal clearance [180]. Cardiac disease can have a significant impact on ochronotic patients and thus if a patient is diagnosed there is a growing consensus for echocardiographic screening after the age of 40 years to detect cardiac involvement and cardiac gated CT to assess coronary artery calcification.
Several case reports have reported different pathologic features associated with ochronotic CM with the involvement of the aorta, coronary vessels and heart valves. The pericardium may be involved in elderly patients [183-186]. In a case report of a 78-year old male patient with known alkaptonuria and severe symptomatic aorta, pathologic examination of the aortic valve and the ascending aorta revealed intracellular and extracellular deposits of ochronotic pigments. The extracellular deposits partially represent degenerated cells appearing as valvular calcifications [183]. In the case of a 55-year old male patients presenting with persistent hip and low-back pain non-responsive to NSAIDs and physiotherapy, laboratory findings reveal normal erythrocyte sedimentation rate (ESR), C-reactive protein (CRP). Heart echocardiography revealed interventricular septum and posterior wall hypertrophy (13.3 mm, 12 mm), thickening of the aortic cusps with calcification, moderate decreased opening and mild aortic insufficiency [185]. The atherosclerotic process is accelerated by the deposition of ochronotic pigment, and a frequent mode of death in patients with alkaptonuria is myocardial infarction [186].
Clinical management: Currently, there is no reliable therapy for ochronosis. Nitisinone, a potent inhibitor of the HGO enzyme that produces HGA, which has shown effectiveness in reducing HGA levels in alkaptonuria [185. In a small trial enrolling 18 patients treated for 3 years with nitisinone. Aortic valve dysfunction (aortic valve velocities > 0.3m/s) was reported in four (4) patents in the treatment arm and one (1) in the control arm. Although the small sample was difficult to draw reliable conclusions but suggest the effect may be maximized if treatment begun early [193]. Other studies report nitisinone-associated marked increase in tyrosine levels that can potentially lead to corneal irritation, dermatological and neurological side effects and long-term effect of nitisinone remains uncertain [179,194]. Attempts to treat alkaptonuria with high dose vitamin C and dietary restriction of tyrosine and phenylamine intake failed to produce a decrease in HGA levels [195]. Currently, the standard therapy consists of a diet low in phenylalanine and tyrosine, and targeted therapy for cardiac symptoms as per the current HF guidelines [180]. Cardiac surgery may be considered in older ochronotic patients (48-67 years) with calcified aortic valves but long-term effect of surgical intervention have not been investigated [180].
A meta-analysis was conducted to evaluate the current diagnostic methods and features for CM due to deposition diseases. Studies of interest were those that assessed patients with known or suspected deposition diseases that can cause CM. Online databases PubMed, Embase and Google Scholar were searched to identify relevant studies. Key words used for the search included deposition diseases (amyloidosis, haemochromatosis, oxalosis and ochronosis), diagnosis or prognosis, (echocardiography, ECG or cardiac MRI. There was no language or publication date restriction. The inclusion criteria were studies evaluating the diagnostic and/or prognostic role of ECG, echocardiography and cardiac MRI in patients with CM due to deposition diseases. The exclusion criteria were studies that enrolled patients with ischemic and non-ischemic CM, acute myocarditis and hypertrophic and infiltrative CM. All abstracts and full-text report of articles meeting the inclusion criteria were retrieved and studied.
Findings: The online search query yielded 109 articles. Of these, only 18 articles that satisfied the inclusion criteria formed the final dataset for this meta-analysis [196-213]. Table 3 provides a summary of study characteristics and main findings. In total, the 18 studies enrolled 1,103 patients with biopsy proven cardiac amyloidosis (CA), older (mean age = 62 years; range = 47-73), and with a greater proportion of male patients based on sixteen studies [196,198-212] having 621:314 male to female ratio translating into 66.4% males. Ten of the studies [196-205] evaluated diagnostic and prognostic value of LGE-CMRI while the remaining eight evaluated the diagnostic value of echocardiography [206-213]. Of the ten CMRI studies, eight were prospective single-centre clinical trials [196,197,199,200,202-205], while the remaining two were retrospective single-centre studies [198,201]. All the CMRI patients had biopsy-proven cardiac amyloidosis and diagnostic LGE images.
Table 3. Characteristics of included studies
Author [Ref #] |
Year |
Study Design |
No. of Patients |
Mean Age (yrs.) |
M/F Ratio |
Diagnostic Tests |
Summary of Key Clinical and Imaging Findings |
Maceira [196] |
2008 |
Prospective single center |
29 |
58±10 |
15/14 |
LGE-CMRI |
Presence of LGE in CMRI provides unique prognostic information relating to disease burden and mortality risk |
Vogelsberg [197] |
2008 |
Prospective single center |
33 |
64±13 |
NA |
LGE-CMRI |
LGE-CMRI can be used to diagnose or rule out CA with good sensitivity and excellent specificity in a clinical setting. |
Austin [198] |
2009 |
Retrospective single center |
47 |
62(51-75) |
33/14 |
LGE-CMRI |
LGE-CMRI pattern is more accurate for diagnosis and a stronger predictor of one-year mortality. |
Migrino [199] |
2009 |
Prospective single center |
29 |
62±11.5 |
15/14 |
LGE-CMRI |
LGE is common and associated with poor long-term prognosis in CA patients and may be useful for risk stratification. |
Ruberg [200] |
2009 |
Prospective single center |
28 |
62±11 |
20/8 |
LGE-CMRI |
LGE is highly sensitive and specific for the identification of cardiac involvement but does not predict survival. |
Mekinian [201] |
2010 |
Retrospective single center |
29 |
63±11 |
19/10 |
LGE-CMRI |
Positive CMRI is associated with a significantly increased risk of death but not independent of clinical HF. |
Aquaro [202] |
2014 |
Prospective single center |
59 |
69±10 |
36/23 |
LGE-CMRI |
LGE-CMRI has a high sensitivity and specificity in diagnosis CA associated with increased risk of death. |
White [203] |
2014 |
Prospective single center |
90 |
62±13 |
52/38 |
LGE-CMRI |
Global hyper-enhancement in CMRI accurately identifies patients with biopsy proven CA and a strong predictor of mortality. |
Fontana [204] |
2015 |
Prospective single center |
250 |
67±12 |
181/69 |
LGE-CMRI |
Transmural LGE shows a continuum of cardiac involvement in CA patients |
Lin [205] |
2018 |
Prospective single center |
82 |
55.5±8.5 |
52/30 |
LGE-CMRI |
Both LGE and ECV are independent prognostic for mortality in CA patients. |
Klein [206] |
1989 |
Prospective single center |
53 |
60±12 |
37/16 |
ECG, Echo - 2D, Doppler |
CA exhibits a spectrum of diastolic filling abnormalities but the restrictive filling pattern is seen only in the advanced disease stages. |
Klein [207] |
1990a |
Prospective single center |
41 |
58±10 |
28/13 |
Echo - 2D, Doppler |
RV diastolic function is abnormal in CA and restrictive filling patterns manifests in the later stages of the disease. |
Klein [208] |
1990b |
Prospective single center |
41 |
59±11 |
36/15 |
Echo - 2D, Doppler |
Serial left ventricular flow velocity variables occur during short-term follow-up evaluation predominantly in the early group with CA |
Tei [209] |
1996 |
Prospective single center |
45 |
60±10 |
29/16 |
Echo - 2D, Doppler |
Doppler-derived index of combined systolic and diastolic dysfunction correlates with global cardiac dysfunction and is a useful predictor of clinical outcomes in CA. |
Dubrey [210] |
1997 |
Prospective single center |
36 |
47±11 |
23/13 |
Echo - 2D, Doppler |
Cardiac involvement in AL and familial CA is indistinguishable but preservation of ECG voltage in familial CA suggest biochemical properties of different types of amyloid fibrils exert different pathological effect, which becomes important in evaluation, management and treatment of different types of CA. |
Ng (SSA) [211] |
2005 |
Prospective single center |
18 |
73±5 |
18/0 |
Serum markers, Echo - 2D, Doppler |
LV wall thickness in AL is greater than in SSA patients but severity of HF in AL is less than in SSA group who have much longer survival. |
Ng (AL) [211] |
2005 |
Prospective single center |
18 |
57±9 |
11/7 |
Pinney (AL) [212] |
2013 |
Prospective single center |
36 |
63 |
NA |
Echo - 2D, Doppler |
Older age at diagnosis and lower NT-proBNP level can help distinguish ATTR from AL CA. Positive troponin T, pacemaker and NYHA class symptoms suggest poor prognosis in ATTR patients. |
Pinney (ATTR) [212] |
2013 |
Prospective single center |
99 |
73 |
NA |
Echo - 2D, Doppler |
Pagourelias [213] |
2017 |
Prospective single center |
40 |
65.5±10.8 |
26/14 |
Echo, Clinical parameters |
Thickened hearts, EF global longitudinal strain ratio has the best accuracy in detecting CA even in patients with mild hypertrophy and preserved EF. |
AL: Light-chain Amyloidosis; ATTR: Amyloidosis Transthyretin; CA: Cardiac Amyloidosis; CMRI: Cardiac Magnetic Resonance Imaging; ECG: Electrocardiogram; Echo: Echocardiography; LGE: Late Gadolinium Enhancement; LV: Left Ventricular; NYHA: New York Heart Association; SSA: Systematic Senile Amyloidosis
In a pooled analysis of eight studies [196,198-201,203-205] evaluating cardiac MRI, 350 surviving patients out of 584 with biopsy proven CA showed the presence of LGE (LGE +ve) translating into an event rate (ER) of 56.5% (95% CI: 42.1 to 69.9: Figure 1). On the other hand, the number of surviving patients without any presence of LGE (LGE –ve) was 158 out of 584 (ER: 28.6%; 95% CI: 16.9 to 44.1: Figure 2). These findings suggest that LGE-CMRI might be a promising method to diagnose or to exclude CA obviating the need for invasive endomyocardial biopsy (EMB). The included CMRI studies also revealed prognostic value of LGE-CMRI. In seven cardiac MRI studies [196,198-201,203,204], there were 147 deaths out of 426 patients. The majority of deaths (n=129; 87.8%) were in patients positive for LGE compared to those negative for LGE (n=18; 12.2%). In addition, pooled analysis of the seven studies revealed a statistically significant four-fold higher odds for death in LGE +ve patients compared to LGE –ve patients (Odds Ratio [OR]: 4.062; 95% CI: 1.340 to 12.313; p=0.013: Figure 3).
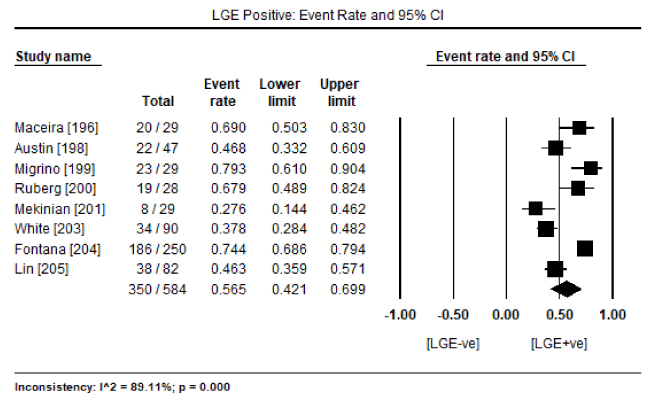
Figure 1. Percentage of biopsy-Proven CA patients with the presence of LGE
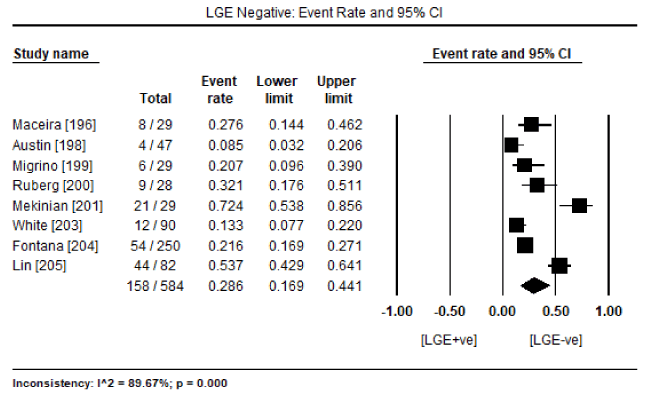
Figure 2. Percentage of biopsy-proven CA patients without the presence of LGE
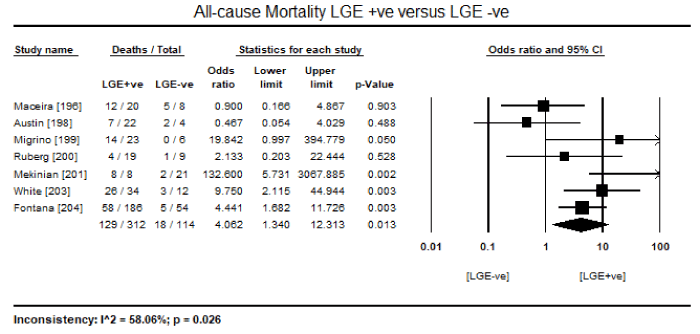
Figure 3. Odds ratio for all-cause mortality in LGE +ve versus LGE –ve patients
Finally, a pooled analysis of five studies [197,198,200,201,203] revealed LGE- cardiac MRI has a high diagnostic sensitivity of 89% (95% CI: 82 to 94; Table 4) and diagnostic specificity of 88% (95% CI: 77 to 94; Table 5). Pooled positive predictive value was 92.1% (95% CI: 88.9 to 94.5%) and negative predictive value was 86.8% (95% CI: 71.7 to 94.5%). The most common ECG abnormality reported in seven studies [198,199,203,206,210,211,214] is low voltage observed in 171 out of 331 patients (event rate: 48.8%; 95 CI: 37.1 to 60.7%;
Table 4. Pooled sensitivity for LGE diagnosis for cardiac amyloidosis
Study (Author) |
Sensitivity (%) |
95% CI |
Vogelsberg [197] |
80 |
71 to 87 |
Austin [198] |
88 |
80 to 93 |
Ruberg [200] |
86 |
77 to 91 |
Mekinian [201] |
98 |
92 to 99 |
White [203] |
93 |
86 to 96 |
Pooled Sensitivity |
89 |
82 to 94 |
Table 5. Pooled specificity for LGE diagnosis for cardiac amyloidosis
Study (Author) |
Specificity (%) |
95% CI |
Vogelsberg [197] |
94 |
87 to 97 |
Austin [198] |
90 |
82 to 94 |
Ruberg [200] |
86 |
77 to 91 |
Mekinian [201] |
93 |
86 to 96 |
White [203] |
70 |
70 to 78 |
Pooled Specificity |
88 |
77 to 94 |
On echocardiography, the most common diagnostic features reported were markers for diastolic dysfunction: mitral inflow deceleration time (DT), peak early to late diastolic velocity (E/A) ratio and mitral annular septal velocity (E/E’ ratio). On echocardiography, patients with CA exhibit shorter DT and higher E/A ratios. In pooled DT data from seven studies [206-210,211,213] enrolling 292 patients showed mean DT of 175 ms (95% CI: 163 to 187 ms) and pooled E/A data from eight studies [206-213] enrolling 427 patients had mean E/A ratio of 1.93 (95% CI: 1.62 to 2.25). Compared with healthy controls, CA patients had significantly shorter mean DT (weighted mean difference [WMD]: -28.16; 95% CI: -55.94 to -0.374; p = 0.047; Figure 5) and a tendency towards a higher E/A ratio (WMD: 0.37; 95% CI: -0.17 to 0.92; p = 0.18; Figure 6) in four studies [206-209]. Significantly more CA patients had E/E’ ratio > 15 and DT < 160 ms compared to healthy controls or LGE +ve patients [198,203]
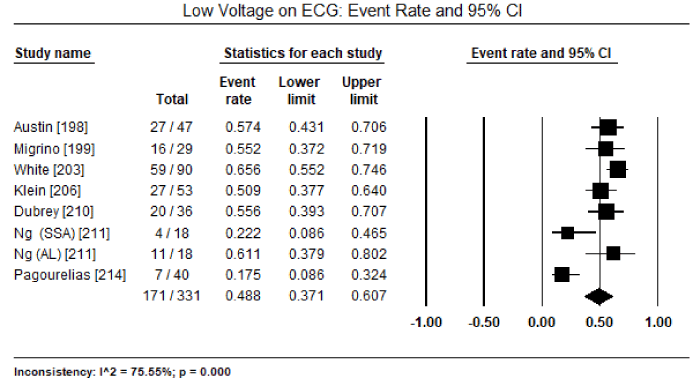
Figure 4. Event rate for low voltage on ECG
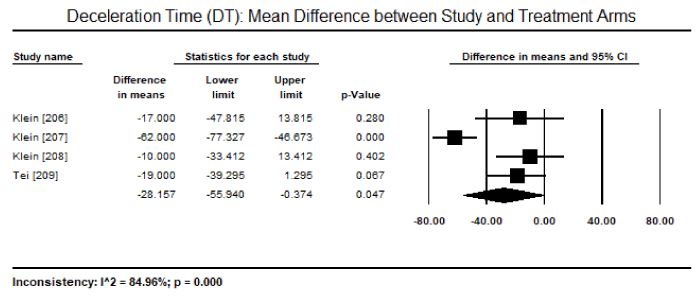
Figure 5. Mean difference in DT between study and treatment groups
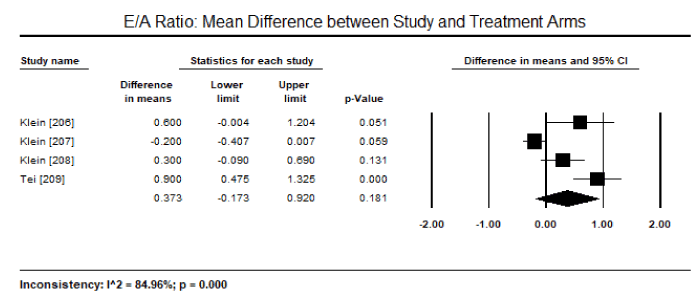
Figure 6. Mean difference in EA ratio between study and treatment groups
The role of cardiac imaging and serum levels of cardiac biomarkers have been well documented both in the diagnosis and prognosis of specific types of CMs, particularly in morphologically classified dilated, hypertrophic and restrictive CMs. However, the role of cardiac imaging in the diagnosis and prognosis of CMs due to deposition diseases remains unclear partly because specific studies on deposition diseases with cardiac involvement are sporadic and largely based on case reports. The present meta-analysis sought to evaluate the diagnosis and prognosis of CM associated with deposition diseases. However, in the online search for studies, it became apparent that except for amyloid CMs, the other forms of CM-associated deposition diseases such as haemochromatosis, oxalosis and ochronosis lack specific large-scale clinical trials. In the end, clinical studies included in this meta-analysis only evaluated patients with CMs due to amyloidosis leading to an incomplete understanding of diagnostic features potentially shared by deposition diseases CMs. Understanding deposition diseases CMs is clinically relevant since patients usually have a poor prognosis, and early diagnosis and prompt institution of treatment is essential to improve clinical outcomes. Despite the limitation of data on deposition disease CM, the findings of the current meta-analysis raises important findings about the diagnostic and prognostic role of echocardiography and cardiac MRI, which is consistent with and supports four previous meta-analyses on CA [214-217] and expert consensus on the diagnosis of deposition disease CM [3].
Echocardiography imaging characteristics
The present meta-analysis reveals that diastolic dysfunction on echocardiography is a common finding in CM due to amyloidosis with cardiac involvement. Although on average all the patients with biopsy proven CA enrolled in the present meta-analysis had preserved or mildly depressed systolic function at baseline, they exhibited a significantly shorter DT (WMD: -28.16; 95% CI: -55.94 to -0.374) and higher peak early to late diastolic velocity (E/A) ratio (WMD: 0.37; 95% CI: -0.17 to 0.92). These findings suggests impaired LV relaxation and pseudonormal or restrictive diastology on echocardiography. These findings are consistent with earlier reports and proposed pathophysiology theories hypothesizing that at early stages of the CM due to deposition diseases, the LV systolic function (LVEF) is generally preserved but gets involved as the disease progresses [62,63]. Although thickened ventricular walls have been described as a key pathophysiology cardiac feature in CA patients, there was no sufficient data for a pooled analysis. However, individual studies [206-208] mentioned bi-ventricular thickening on echocardiography. Thickened ventricular walls have been implicated as a cause of non-dilated (stiff and poorly compliant) ventricles resulting in progressive abnormalities in diastolic filling [3]. While systolic function is often preserved or mildly depressed on early stages of CM associated with CA, it is a less reliable diagnostic feature due to reduced end-diastolic volume producing a low stroke volume [41-43]. In addition to diastolic abnormalities on Doppler echocardiography, the present findings are consistent with recommendations of the AHA on the use of echocardiography for detecting thickened bi-ventricular walls, impaired relaxation and restrictive filling for the diagnosis of amyloid CM [3]. In particular, thickened bi-ventricular walls not explained by known causes such as hypertension and aortic stenosis raises the clinical suspicion of CM due to deposition diseases with cardiac involvement [3].
LGE diagnosis and prognosis: Cardiac MRI remains an option in the diagnosis of CM due to deposition diseases often considered after ECG, echocardiography and/or EMB. The present meta-analysis reveals LGE-CMRI has important diagnostic and prognostic value in patients with CM associated with deposition disease (CA). More than half of biopsy proven patients (56%) show the presence of LGE on CMRI, which has a high diagnostic sensitivity (89%), specificity (88%), positive predictive value (92%) and negative predictive value (87%) in reference to EMB. These findings suggest LGE-CMRI has a promising value in diagnosing or excluding CM due to CA as well as obviating the need for EMB. In these patients, LGE-CMRI also has an important prognostic value. The presence of LGE is associated with a four-fold higher risk of death compared to LGE negative patients. Consistent with these findings, a previous meta-analysis [214] describes CMRI as having a diagnostic advantage in detecting cardiac deposition using LGE to detect and quantify deposition in patients with biopsy proven CA. In prior systematic reviews [212-214], CMRI appears a reasonable alternative to EMB in patients with tissue diagnosis from an alternative sight and thought to be at high risk for invasive investigation. The three reviews also suggest LGE is a marker of increased mortality irrespective of cardiac biopsy results. Although LGE has emerged as a gold standard CMRI modality in evaluating CA, other newer techniques such as T1-mapping, intra myocardial T1 difference, loon locker, T1 scout and equilibrium contrast-enhanced CMRI has been successful in characterizing diffuse myocardial fibrosis [214,217]. There is need for further evaluation of these newer CMRI techniques in the diagnosis and prognosis of other deposition diseases.
Clinical implications: Cardiomyopathy associated with deposition diseases often has a poor prognosis. Thus, better diagnostic tests might permit earlier detection and targeted intervention to improve clinical and patient outcomes [3]. At present, the most common methods used in the diagnosis of CMs are ECG and echocardiography due to widespread availability, ease of use and convenience. However, both methods have little use in CMs due to deposition diseases because of low detection accuracy in the early stages of the disease as well as lack pathognomonic features to confirm diagnosis in clinical practice that EMB is not in common use [214]. ECG evaluation is unsuitable for patients with pulmonary disorders yet many elderly CM patients are more likely to have cardiopulmonary involvement. Echocardiography on the other hand can only detect advanced cases but cannot easily assess earlier cases or differentiate certain deposition diseases such as CA from HCM because both have thickened ventricular walls with speckled myocardium [6,218].
Although EMB is a gold standard in diagnosis of some deposition diseases such as CA because deposition occurs throughout the myocardial tissue [33-36], it is less useful in some deposition diseases such as haemochromatosis due to patchy deposition of iron on the myocardial tissues leading to many cases of false positive findings [134]. In addition, EMB has limited used in clinical settings due to unavailability, relatively high risks and clinical complications [214]. LGE-CMRI is thus emerging as an important non-invasive imaging technique to detect and quantify cardiac deposition to support diagnosis as well as acts as an independent diagnostic marker with a promise to obviate the need for invasive EMB in certain deposition diseases with cardiac involvement. However, there is need for further clinical trials to confirm or identify other newer non-invasive imaging features to improve early diagnosis of CM due to deposition diseases to inform targeted treatment and improve clinical and patient outcomes.
Deposition diseases amyloidosis, haemochromatosis, oxalosis and ochronosis cause CM as a consequence of the accumulation of amyloid fibrils, iron, calcium oxalate and homogentisic acid respectively in the myocardial tissues. Clinical presentation and diagnosis varies depending on the type of substance deposited, location and stage of the disease. Amyloid CM initially manifests as progressive diastolic dysfunction, and in the later stages of the disease, as biventricular dysfunction often accompanied by conduction disturbance. EMB remains the diagnostic gold standard while LGE-CMRI is an independent prognostic marker for mortality. Hemochromatosis initially manifests as restrictive hemodynamics and increased filling pressure, and then progresses into systolic dysfunction and bi-ventricular dilation. Cardiac MRI is the only non-invasive modality used to detect and quantify iron deposition in hemochromatosis patients while patchy iron deposits on the myocardium limits the diagnostic accuracy and the utility of EMB in clinical practice. Oxalosis principally manifests as kidney dysfunction with sinus tachycardia, AV block progressing into heart block, mildly dilated LV with moderately to severely depressed systolic function, bi-ventricular concentric thickening with patchy echodense speckled myocardial reflections. Cardiac involvement is uncommon in ochronosis often manifesting as the calcification of aorta, coronary vessels and heart valves. Treatment usually targets the underlying disease: inhibiting amyloid fibril formation (amyloid), phlebotomy (haemochromatosis), vitamins and mineral therapy, and alkalization of urine (oxalosis) and nitisinone (ochronosis).
- Touart DM, Sau P (1998) Cutaneous deposition diseases. Part I. J Am Acad Dermatol 39: 149-171. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. (2006) Contemporary definitions an classification of the cardiomyopathies: an American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113: 1807-1816. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Donnelly JP, Hanna MA (2017) Cardiac amyloidosis: An update on diagnosis and treatment. Cleve Clin J Med 84: 12-26. [Crossref]
- Liao R, Ward JE (2017) Amyloid Cardiomyopathy: Disease on the Rise. Circ Res 120: 1865-1867. [Crossref]
- Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, et al. (2013) Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis 106: 528-540. [Crossref]
- Garcia-Pavia P, Tome-Esteban MT, Rapezzi C (2011) Amyloidosis. Also a heart disease. Rev Esp Cardiol 64: 797-808. [Crossref]
- Falk RH (2005) Diagnosis and management of the cardiac amyloidoses. Circulation 112: 2047-2060. [Crossref]
- Giraudeau C, Babuty D, Goupille P (2000) Pericardial effusion revealing cardiac amyloidosis in the course of rheumatoid arthritis. Arch Mal Coeur Vaiss 93: 1145-1149. [Crossref]
- Bunker D, Gorevic P (2012) AA amyloidosis: Mount Sinai experience, 1997-2012. Mt Sinai J Med 79: 749-756. [Crossref]
- Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD (2012) Updates in cardiac amyloidosis: a review. J Am Heart Assoc 1: e000364. [Crossref]
- Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, et al. (1992) Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 79: 1817-1822. [Crossref]
- Krsnik I, Cabero M, Morillo D, Segovia J, García-Pavía P, et al. (2015) Light chain amyloidosis: Experience in a tertiary hospital: 2005-2013. Rev Clin Esp 215: 1-8. [Crossref]
- Kyle RA, Gertz MA (1995) Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 32: 45-59. [Crossref]
- Merlini G, Stone MJ (2006) Dangerous small B-cell clones. Blood 108: 2520-2530. [Crossref]
- Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, et al. (2004) Serum cardiac troponins and N-terminal pro–brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 22: 3751-3757. [Crossref]
- Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, et al. (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med 336: 466-473. [Crossref]
- Falk RH (2002) The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis 12: 141-143. [Crossref]
- Andrade C (1952) A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75: 408-427. [Crossref]
- Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349: 583-596. [Crossref]
- Gioeva Z, Urban P, Rüdiger Meliss R, Haag J, Axmann HD, et al. (2013) ATTR amyloid in the carpal tunnel ligament is frequently of wildtype transthyretin origin. Amyloid 20: 1-6. [Crossref]
- Yanagisawa A, Ueda M, Sueyoshi T, Okada T, Fujimoto T, et al. (2015) Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol 28: 201-207. [Crossref]
- Siddiqi OK, Ruberg FL (2018) Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med 28: 10-21. [Crossref]
- Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, et al. (2016) Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 23: 58-63. [Crossref]
- Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, et al. (2008) Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med 40: 232-239. [Crossref]
- González-López E, Gallego-Delgado M, Guzzo-Merello G, de Haro-del Moral FJ, Cobo-Marcos M, et al. (2015) Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 36: 2585-2594. [Crossref]
- Ogiwara F, Koyama J, Ikeda S, Kinoshita O, Falk RH (2005) Comparison of the strain Doppler echocardiographic features of familial amyloid polyneuropathy (FAP) and light-chain amyloidosis. Am J Cardiol 95: 538-540. [Crossref]
- Maleszewski JJ (2015) Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol 24: 343-350. [Crossref]
- Wright JR, Calkins E (1975) Amyloid in the aged heart: frequency and clinical significance. J Am Geriatr Soc 23: 97-103. [Crossref]
- Vermeer AMC, Janssen A, Boorsma PC, Mannens MMAM, Wilde AAM, et al. (2017) Transthyretin amyloidosis: a phenocopy of hypertrophic cardiomyopathy. Amyloid 24 :87-91. [Crossref]
- Velazquez-Ceceña JL, Lubell DL, Nagajothi N, Al-Masri H, Siddiqui M, et al. (2009) Syncope cardiomyopathy in a patient with primary AL-type amyloid heart disease. Tex Heart Inst J 36: 50-54. [Crossref]
- Stegman BM, Kwon D, Rodriguez ER, Hanna M, Cho L (2014) Left ventricular hypertrophy in a runner: things are not always what they seem. Circulation 130: 590-592. [Crossref]
- Ardehali H, Qasim A, Cappola T (2004) Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 147: 919-923. [Crossref]
- Berghoff M, Kathpal M, Khan F, Skinner M, Falk R, et al. (2003) Endothelial dysfunction precedes C-fi ber abnormalities in primary (AL) amyloidosis. Ann Neurol 53: 725-730. [Crossref]
- Khan MF, Falk RH (2001) Amyloidosis. Postgrad Med J 77: 686-693. [Crossref]
- Martinez-Naharro A, Treibel TA, Abdel-Gadir A (2017) Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 70: 466-477. [Crossref]
- Longhi S, Quarta CC, Milandri A (2015) Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid 22: 147-155. [Crossref]
- Sperry BW, Vranian MN, Hachamovitch R (2016) Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol 214: 477-481. [Crossref]
- Berk JL, Keane J, Seldin DC (2003) Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 124: 969-977. [Crossref]
- Miani D, Rocco M, Alberti E, Spedicato L, Fioretti PM (2002) Amyloidosis of epicardial and intramural coronary arteries as an unusual cause of myocardial infarction and refractory angina pectoris. Ital Heart J 3: 479-482. [Crossref]
- Mesquita ET, Jorge AJL, Souza CV Junior, Andrade TR (2017) Cardiac amyloidosis and its new clinical phenotype: heart failure with preserved ejection fraction. Arg Bras Cardiol 109: 71-80. [Crossref]
- Swanton RH, Brooksby IAB, Davies MJ, Coltart DJ, Jenkins BS, et al. (1977) Systolic and diastolic ventricular function in cardiac amyloidosis: studies in six cases diagnosed with endomyocardial biopsy. Am J Cardiol 39: 658-664. [Crossref]
- Tyberg TI, Goodyer AVN, Hurst VW 3rd, Alexander J, Langou RA (1981) Left ventricular filling in differentiating restrictive amyloid cardiomyopathy and constrictive pericarditis. Am J Cardiol 47: 791-796. [Crossref]
- Falk RH, Dubrey SW (2010) Amyloid heart disease. Prog Cardiovasc Dis 52: 347-361. [Crossref]
- Brodarick S, Paine R, Higa E, Carmichael KA (1982) Pericardial tamponade, a new complication of amyloid heart disease. Am J Med 73: 133-135. [Crossref]
- Navarro JF, Rivera M, Ortuno J (1992) Cardiac tamponade as presentation of systemic amyloidosis. Int J Cardiol 36: 107-108 [Crossref]
- Chamarthi B, Dubrey SW, Cha K, Skinner M, Falk RH (1997) Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol 80: 1242-1245. [Crossref]
- Padfield GJ, Maclay JD (2010) Macroglossia and complete heart block in a woman with multiple myeloma. QJM 103: 271-272. [Crossref]
- Payne CE, Usher BW (2007) Atrioventicular block in familial amyloidosis: revisiting an old debate. J SC Med Assoc 103: 119-122. [Crossref]
- Ridolfi RL, Bulkley BH, Hutchins GM (1977) The conduction system in cardiac amyloidosis: clinical and pathologic features of 23 patients. Am J Med 62: 677-686. [Crossref]
- Velazquez-Cecena JL, Lubell DL, Nagajothi N, Al-Masri H, Siddiqui M, et al. (2009) Syncope from dynamic left ventricular outflow tract obstruction simulating hypertrophic cardiomyopathy in a patient with primary AL-type amyloid heart disease. Tex Heart Inst J 36: 50-54. [Crossref]
- Dinwoodey DL, Skinner M, Maron MS, Davidoff R, Ruberg FL (2008) Light-chain amyloidosis with echocardiographic features of hypertrophic cardiomyopathy. Am J Cardiol 101: 674-676. [Crossref]
- Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, et al. (1998) The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 91: 141-157. [Crossref]
- Morner S, Hellman U, Suhr OB, Kazzam E, Waldenstrom A (2005) Amyloid heart disease mimicking hypertrophic cardiomyopathy. J Intern Med 258: 225-230. [Crossref]
- Mueller PS, Edwards WD, Gertz MA (2000) Symptomatic ischemic heart disease resulting from obstructive intramural coronary amyloidosis. Am J Med 109: 181-188. [Crossref]
- AlSuwaidi J, Velianou JL, Gertz MA, Cannon RO, Higano ST, et al. (1999) Systemic amyloidosis presenting with angina pectoris. Ann Intern Med 131: 838-841. [Crossref]
- Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, et al. (2007) Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation 116: 2420-2426. [Crossref]
- Zubkov AY, Rabinstein AA, Dispenzieri A, Wijdicks EF (2007) Primary systemic amyloidosis with ischemic stroke as a presenting complication. Neurology 69: 1136-1141. [Crossref]
- Poels MM, Ikram MA, van der Lugt A, Hofman A, Krestin GP, et al. (2011) Incidence of cerebral microbleeds in the general population: the Rotterdam Scan Study. Stroke 42: 656-661. [Crossref]
- Dubrey SW, Cha K, Anderson J (1998) The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 91:141-157. [Crossref]
- Sperry BW, Jones BM, Vranian MN, Hanna M, Jaber WA (2016) Recognizing transthyretin cardiac amyloidosis in patients with aortic stenosis: impact on prognosis. JACC Cardiovasc Imaging 9: 904-906. [Crossref]
- Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR (2011) Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med 124: 1006-1015. [Crossref]
- Dubrey SW, Hawkins PN, Falk RH (2011) Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart 97: 75-84. [Crossref]
- Simons M, Isner JM (1992) Assessment of relative sensitivities of non-invasive tests for cardiac amyloidosis in documented cardiac amyloidosis. Am J Cardiol 69: 425-427. [Crossref]
- Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, et al. (2005) Noninvasive etiologic diagnosis of cardiac amyloidosis using Tc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 46: 1076-1084. [Crossref]
- Kwong RY, Falk RH (2005) Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 111: 122-124. [Crossref]
- Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, et al. (2005) Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 111: 186-193. [Crossref]
- Nordlinger M, Magnani B, Skinner M, Falk RH (2005) Is elevated plasma B-natriuretic peptide in amyloidosis simply a function of the presence of heart failure? Am J Cardiol 96: 982-984. [Crossref]
- Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, et al. (2003) Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 107: 2440-2445. [Crossref]
- Wechalekar AD, Gillmore JD, Wassef N, Lachmann HJ, Whelan C, et al. (2011) Abnormal N-terminal fragment of brain natriureticpeptide in patients with light chain amyloidosis without cardiac involvement at presentation is a risk factor for development of cardiac amyloidosis. Haematologica 96: 1079-1080. [Crossref]
- Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, et al. (2007) Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood 110: 787-788. [Crossref]
- Ardehali H, Qasim A, Cappola T, Howard D, Hruban R, et al. (2004) Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 147: 919-923. [Crossref]
- Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR 3rd, et al. (2009) Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 114: 4957-4959. [Crossref]
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, et al. (2004) Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol 79: 319-328. [Crossref]
- Yamaguchi T (2008) Syncope and sinus bradycardia from combined use of thalidomide and beta-blocker. Pharmacoepidemiol Drug Saf 17: 1033-1035. [Crossref]
- Rubinow A, Skinner M, Cohen AS (1981) Digoxin sensitivity in amyloid cardiomyopathy. Circulation 63: 1285-1288. [Crossref]
- Gertz MA, Skinner M, Connors LH, Falk RH, Cohen AS, et al. (1985) Selective binding of nifedipine to amyloid fibrils. Am J Cardiol 55: 1646. [Crossref]
- Pollak A, Falk RH (1993) Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest 104: 618-620. [Crossref]
- Low PA (2002) Autonomic neuropathies. Curr Opin Neurol 15: 605-609. [Crossref]
- Kristen AV, Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, et al. (2008) Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm 5: 235-240. [Crossref]
- Bellavia D, Pellikka PA, Abraham TP, Al-Zahrani GB, Dispenzieri A, et al. (2009) ‘Hypersynchronisation’ by tissue velocity imaging in patients with cardiac amyloidosis. Heart 95: 234-240. [Crossref]
- Dhodapkar MV, Hussein MA, Rasmussen E, Solomon A, Larson RA, et al. (2004) Clinical efficacy of high-dose dexamethasone with maintenance dexamethasone/alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood 104: 3520-3526. [Crossref]
- Cohen AD, Zhou P, Chou J, Teruya-Feldstein J, Reich L, et al. (2007) Risk-adapted autologous stem cell transplantation with adjuvant dexamethasone +/- thalidomide for systemic light-chain amyloidosis: results of a phase II trial. Br J Haematol 139: 224-233. [Crossref]
- Comenzo RL (2005) Managing systemic light-chain amyloidosis. J Natl Compr Canc Netw 5: 179-187. [Crossref]
- Wechalekar AD, Hawkins PN, Gillmore JD (2008) Perspectives in treatment of AL amyloidosis. Br J Haematol 140: 365-377. [Crossref]
- Benson MD, Kluve-Beckerman B, Zeldenrust SR, Siesky AM, Bodenmiller DM, et al. (2006) Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve 33: 609-618. [Crossref]
- Sekijima Y, Dendle MA, Kelly JW (2006) Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 13: 236-249. [Crossref]
- Tojo K, Sekijima Y, Kelly JW, Ikeda S (2006) Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res 56: 441-449. [Crossref]
- Kamran SH, Saleem U, Ahmad B, Ahmad M (2012) Effect of iron overload cardiomyopathy in haemochromatosis and β-thalassemia. J Appl Pharm 21: 556-566.
- Aronow WS (2018) Management of cardiac hemochromatosis. Arch Med Sci 14: 560-568. [Crossref]
- Fix OK, Kowdley KV (2008) Hereditary hemochromatosis. Minerva Med 99:605-617. [Crossref]
- Fowler C (2008) Hereditary hemochromatosis: pathophysiology, diagnosis, and management. Crit Care Nurs Clin North Am 20: 191-201. [Crossref]
- Pietrangelo A (2004) Hereditary hemochromatosis: a new look at an old disease. New Eng J Med 350: 2383-2397. [Crossref]
- Kremastinos DT, Farmakis D (2011) Iron overload cardiomyopathy in clinical practice. Circulation 124: 2253-2263. [Crossref]
- Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y (2010) Iron overload cardiomyopathy: better understanding of an increasing disorder. J Am Coll Cardiol 56: 1001-112. [Crossref]
- Pietrangelo A (2004) Hereditary hemochromatosis–a new look at an old disease. N Engl J Med 350: 2383-2397. [Crossref]
- Wood JC (2008) Cardiac iron across different transfusion-dependent diseases. Blood Rev 22: S14-S21. [Crossref]
- Porter JB (2007) Concepts and goals in the management of transfusional iron overload. Am J Hematol 82: 1136-1139. [Crossref]
- Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, et al. (2003) L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 9: 1187-1194. [Crossref]
- Carpenter JP, He T, Kirk P, Roughton M, Anderson LJ, et al. (2011) On T2* magnetic resonance and cardiac iron. Circulation 123: 1519-1528. [Crossref]
- Horwitz LD, Rosenthal EA (1999) Iron-mediated cardiovascular injury. Vasc Med 4: 93-99. [Crossref]
- Liu P, Olivieri N (1994) Iron overload cardiomyopathies: new insights into an old disease. Cardiovasc Drugs Ther 8: 101-110. [Crossref]
- Murphy CJ, Oudit GY (2010) Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Card Fail 16: 888-900. [Crossref]
- Kremastinos DT, Farmakis D, Aessopos A, Hahalis G, Hamodraka E, et al. (2010) Beta-thalassemia cardiomyopathy: history, present considerations, and future perspectives. Circ Heart Fail 3: 451-458. [Crossref]
- Kremastinos DT, Toutouzas PK, Vyssoulis GP, Venetis CA, Avgoustakis DG (1984) Iron overload and left ventricular performance in beta thalassemia. Acta Cardiol 39: 29-40. [Crossref]
- Aessopos A, Berdoukas V, Tsironi M (2008) The heart in transfusion dependent homozygous thalassaemia today–prediction, prevention and management. Eur J Haematol 80: 93-106. [Crossref]
- Farmakis D, Giakoumis A, Polymeropoulos E, Aessopos A (2003) Pathogenetic aspects of immune deficiency associated with beta-thalassemia. Med Sci Monit 9: RA19-RA22. [Crossref]
- Kremastinos DT, Toutouzas PK, Vyssoulis GP, Venetis CA, Vretou HP, et al. (1985) Global and segmental left ventricular function in betathalassemia. Cardiology 72: 129-139. [Crossref]
- Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y (2010) Iron overload cardiomyopathy: better understanding of an increasing disorder. J Am Coll Cardiol 56: 1001-1012. [Crossref]
- Kremastinos DT, Tiniakos G, Theodorakis GN, Katritsis DG, Toutouzas PK (1995) Myocarditis in β-thalassemia major: a cause of heart failure. Circulation 91: 66-71. [Crossref]
- Jessup M, Manno CS (1998) Diagnosis and management of iron-induced heart disease in Cooley’s anaemia. Ann N Y Acad Sci 850: 242-250. [Crossref]
- Kremastinos DT, Tsiapras DP, Kostopoulou AG, Hamodraka ES, Chaidaroglou AS, et al. (2007) NT-proBNP levels and diastolic dysfunction in beta-thalassaemia major patients. Eur J Heart Fail 9: 531-536. [Crossref]
- Kremastinos DT, Hamodraka E, Parissis J, Tsiapras D, Dima K, et al. (2010) Predictive value of B-type natriuretic peptides in detecting latent left ventricular diastolic dysfunction in beta-thalassemia major. Am Heart J 159: 68-74. [Crossref]
- Kremastinos DT (2001) Heart failure in beta-thalassemia. Congest Heart Fail 7: 312-314. [Crossref]
- Kremastinos DT, Tsiapras DP, Tsetsos GA, Rentoukas EI, Vretou HP, et al. (1993) Left ventricular diastolic Doppler characteristics in betathalassemia major. Circulation. 88: 1127-1135. [Crossref]
- Skinner C, Kenmure AC. (1973) Haemochromatosis presenting as congestive cardiomyopathy and responding to venesection. Br Heart J 1973; 35: 466-468. [Crossref]
- Cascales A, Sanchez-Vega B, Navarro N (2012) Clinical and genetic determinants of anthracycline-induced cardiac iron accumulation. Int J Cardiol 2012; 154: 282-286. [Crossref]
- Furth PA, Futterweit W, Gorlin R (1985) Refractory biventricular heart failure in secondary hemochromatosis. Am J Med Sci 290: 209-213. [Crossref]
- Wasserman AJ, Richardson DW, Baird CL (1962) Cardiac hemochromatosis simulating constrictive pericarditis. Am J Med 32: 316-323. [Crossref]
- Feely J, Counihan TB (1977) Haemochromatosis presenting as angina and responding to venesection. Br Med J 2: 681-682. [Crossref]
- Aronow WS, Meister L, Kent JR (1969) Atrioventricular block in familial hemochromatosis treated by permanent synchronous pacemaker. Arch Intern Med 123: 433-435. [Crossref]
- Wu VC, Huang JW, Wu MS (2004) The effect of iron stores on corrected QT dispersion in patients undergoing peritoneal dialysis. Am J Kidney Dis 44: 720-728. [Crossref]
- Rose RA, Sellan M, Simpson JA (2011) Iron overload decreases CaV1.3-dependent L-type Ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ Arrhythm Electrophysiol 118: 174-177. [Crossref]
- Klintschar M, Stiller D (2004) Sudden cardiac death in hereditary hemochromatosis: an underestimated cause of death? Int J Legal Med 118: 174-177. [Crossref]
- Campbell S, George DK, Robb SD (2003) The prevalence of haemochromatosis gene mutations in the West of Scotland and their relation to ischaemic heart disease. Heart 89: 1023-1026. [Crossref]
- Candore G, Balistreri CR, Lio D (2003) Association between HFE mutations and acute myocardial infarction: a study in patients from Northern and Southern Italy. Blood Cells Mol Dis 31: 57-62. [Crossref]
- Schmitt B, Golub RM, Green R (2005) Screening primary care patients for hereditary hemochromatosis with transferrin saturation and serum ferritin level: systematic review for the American College of Physicians. Ann Intern Med 143: 522-536. [Crossref]
- Qaseem A, Aronson M, Fitterman N, Snow V, Weiss KB, et al. (2005) Screening for hereditary hemochromatosis: a clinical practice guideline from the American College of Physicians. Ann Intern Med 143: 517-521. [Crossref]
- Beutler E, Felitti V, Gelbart T, Ho N (2000) The effect of HFE genotypes on measurements of iron overload in patients attending a health appraisal clinic. Ann Intern Med 133: 329-337. [Crossref]
- Harris EL, McLaren CE, Reboussin DM (2007) Serum ferritin and transferrin saturation in Asians and Pacific Islanders. Arch Intern Med 167: 722-726. [Crossref]
- Lubitz SA, Goldbarg SH, Mehta D (2008) Sudden cardiac death in infiltrative cardiomyopathies: sarcoidosis, scleroderma, amyloidosis, hemochromatosis. Prog Cardiovasc Dis 51: 58-73. [Crossref]
- Palka P, Macdonald G, Lange A, Burstow DJ (2002) The role of Doppler left ventricular filling indexes and Doppler tissue echocardiography in the assessment of cardiac involvement in hereditary hemochromatosis. J Am Soc Echocardiogr 15: 884-890. [Crossref]
- Palka P, Lange A, Atherton J, Stafford WJ, Burstow DJ (2004) Biventricular diastolic behaviour in patients with hypertrophic and hereditary hemochromatosis cardiomyopathies. Eur J Echocardiogr 5: 356-366. [Crossref]
- Buja LM, Roberts WC (1971) Iron in the heart. Aetiology and clinical significance. Am J Med 51: 209-221. [Crossref]
- Rivers J, Garrahy P, Robinson W (1987) Reversible cardiac dysfunction in hemochromatosis. Am Heart J 113: 216-217. [Crossref]
- Easley RM Jr, Screiner BF Jr, Yu PN (1972) Reversible cardiomyopathy associated with hemochromatosis. N Engl J Med 287: 866-867. [Crossref]
- Niederau C, Fischer R, Sonnenberg A (1985) Survival and causes of death in cirrhotic and in non-cirrhotic patients with primary hemochromatosis. N Engl J Med 313: 1256-1262. [Crossref]
- Rahko PS, Salerni R, Uretsky BF (1986) Successful reversal by chelation therapy of congestive cardiomyopathy due to iron overload. J Am Coll Cardiol 8: 436-840. [Crossref]
- Barton JC, McDonnell SM, Adams PC (1998) Management of hemochromatosis. Ann Intern Med 129: 932-939. [Crossref]
- Jomova K, Valko M (2011) Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des 17: 3460-3473. [Crossref]
- Crosby WH (1986) Hemochromatosis. Treatment to alleviate injury. Arch Intern Med 146: 1910-1911. [Crossref]
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS (2011) American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54: 328-343. [Crossref]
- Adams PC, Barton JC (2010) How I treat hemochromatosis. Blood 116: 317-325. [Crossref]
- Pietrangelo A (2005) Non-HFE hemachromatosis. Hepatology 39: 21-29. [Crossref]
- McLaran CJ, Bett JH, Nye JA (1982) Congestive cardiomyopathy and haemochromatosis-rapid progression possibly accelerated by excessive ingestion of ascorbic acid. Aust N Z J Med 12: 187-188. [Crossref]
- Herbert V (1999) Hemochromatosis and vitamin C. Ann Intern Med 131: 475-476. [Crossref]
- Milward EA, Baines SK, Knuiman MW (2008) Noncitrus fruits as novel dietary environmental modifiers of iron stores in people with or without HFE gene mutations. Mayo Clin Proc 83: 543-549. [Crossref]
- Conrad ME, Barton JC (1980) Anaemia and iron kinetics in alcoholism. Semin Hematol 17: 149-163. [Crossref]
- Celada A, Rudolph H, Donath A (1979) Effect of experimental chronic alcohol ingestion and folic acid deficiency on iron absorption. Blood 54: 906-915. [Crossref]
- Yancy CW, Jessup M, Bozkurt B (2013) 2013 ACCF/ AHA guidelines for the management of heart failure: executive summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American College of Chest Physicians, Heart Rhythm Society, and International Society for Heart and Lung Transplantation. Endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation. J Am Coll Cardiol 62: 1495539. [Crossref]
- Dabestani A, Child JS, Henze E (1984) Primary hemochromatosis: anatomic and physiologic characteristics of the cardiac ventricles and their response to phlebotomy. Am J Cardiol 54: 153-159. [Crossref]
- Pennell DJ, Udelson JE, Arai AE (2013) Cardiovascular function and treatment in beta-thalassemia major: a consensus statement from the American Heart Association. Circulation 128: 281-308. [Crossref]
- Hughes DT (1959) The clinical and pathological background of two cases of oxalosis. J Clin Pathol 12: 498-509. [Crossref]
- Chou LY, Donohue WL (1952) Oxalosis: possible" inborn error of metabolism" with nephrolithiasis and nephrocalcinosis due to calcium oxalate as the predominating features. Pediatrics 10: 660-666. [Crossref]
- Scowen EF, Stansfeld AG, Watts RW (1959) Oxalosis and primary hyperoxaluria. J Pathol Bacteriol 77: 195-205. [Crossref]
- Velez-Roa S, Depierreux M, Nortier J, Unger P (2006) Cardiac oxalosis: a rare cause of diastolic dysfunction. Eur Heart J 27: 2496-. [Crossref]
- Detry O, Honoré P, DeRoover A, Trimeche M, Demoulin JC, et al. (2002) Reversal of oxalosis cardiomyopathy after combined liver and kidney transplantation. Transpl Int 15: 50-52. [Crossref]
- Von Unruh GE, Voss S, Sauerbruch T (2004) Dependence of oxalate absorption on the daily calcium intake. J Am Soc Nephro 15: 1567-1573. [Crossref]
- Nazzal L, Puri S, Goldfarb DS (2015) Enteric hyperoxaluria: an important cause of end-stage kidney disease. Nephrol Dial Transplant 31: 375-382. [Crossref]
- Palka P, Duhig E, Carey L, Galbraith A (2001) Primary oxalosis with cardiac involvement: echocardiographic features of an unusual form of cardiomyopathy. Circulation 103: e122-123. [Crossref]
- Mookadam F, Smith T, Jiamsripong P, Moustafa SE, Monico CG, et al. (2010) Cardiac abnormalities in primary hyperoxaluria. Circ J 74: 2403-2409. [Crossref]
- Coltart DJ, Hudson RE (1971) Primary oxalosis of the heart: a cause of heart block. Br Heart J 33: 315. [Crossref]
- Yaich S, Chaabouni Y, Charfeddine K, Zaghdane S, Kharrat M, et al. (2014) Secondary oxalosis due to excess vitamin C intake: a cause of graft loss in a renal transplant recipient. Saudi J Kidney Dis Transpl 25: 113-116. [Crossref]
- Pinto YM, Elliott PM, Arbustin E, Adler Y, Anastasakis A, et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37: 1850-1858. [Crossref]
- Maron BJ, Udelson JE, Bonow RO, Nishimura RA, Ackerman MJ, et al. (2015) Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 3: hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientific statement from the American Heart Association and American College of Cardiology. Circulation 132: e273-e280 [Crossref]
- Rumsby G, Williams E, Coulter-Mackie M (2004) Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int 66: 959-963. [Crossref]
- Hoppe B, Kemper MJ, Bokenkamp A, Langman CB (1998) Plasma calcium-oxalate saturation in children with renal insufficiency and in children with primary hyperoxaluria. Kidney Int 54: 921-925. [Crossref]
- Langman CB (2001) The optimal approach to the patient with oxalosis. Adv Ren Replace Ther 8: 214-222. [Crossref]
- Yoshioka J, Park YD, Tanaka Y, Kobayashi Y, Miyajima M, et al. (2001) Echocardiographic features in a patient with primary oxalosis. Echocardiography 18: 599-602. [Crossref]
- Van Driessche L, Dhondt A, De Sutter J (2007) Heart failure with mitral valve regurgitation due to primary hyperoxaluria type I. Case report with review of the literature. Acta Cardiol 62: 202-206. [Crossref]
- Schulze MR, Wachter R, Schmeisser A, Fischer R, Strasser RH (2006) Restrictive cardiomyopathy in a patient with primary hyperoxaluria type II. Clin Res Cardiol 95: 235-240. [Crossref]
- Tonkin AM, Mond HG, Mathew TH, Sloman JG (1976) Primary oxalosis with myocardial involvement and heart block. Med J Aust 1: 873-874. [Crossref]
- West RR, Salyer WR, Hutchins GM (1973) Adult-onset primary oxalosis with complete heart block. Johns Hopkins Med J 133: 195-200. [Crossref]
- Massie BM, Bharati S, Scheinman MM, Lev M, Desai J, et al. (1981) Primary oxalosis with pan-conduction cardiac disease: Electrophysiologic and anatomic correlation. Circulation 64: 845-852. [Crossref]
- Hamaya K, Ohishi K (1981) Primary oxalosis with cardiac manifestations. Acta Pathol Jpn 30: 451-458. [Crossref]
- Quan KJ, Biblo LA (2003) Type 1 primary hyperoxaluria: An unusual presentation of ventricular tachycardia. Cardiol Rev 11: 318-319. [Crossref]
- Lorenz EC, Michet CJ, Milliner DS, Lieske JC (2013) Update on oxalate crystal disease. Curr Rheumatol Rep 15: 340. [Crossref]
- Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, et al. (2012) Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 27: 1729-1736. [Crossref]
- Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, et al. (2002). Natural history of alkaptonuria. N Engl J Med 347: 2111-2121. [Crossref]
- Karavaggelis A, Young C, Attia R (2017) Black heart at surgeryprimary diagnosis of alkaptonuria at surgery. J Cardiol Curr Res 9: 00335.
- Collins EJ, Hand R (2005) Alkaptonuric ochronosis: a case report. AANA J 73: 41-46. [Crossref]
- Gottschalk BH, Blankenstein J, Guo L (2018) Ochronosis of mitral valve and coronary arteries. Ann Thorac Surg 106: e19-20. [Crossref]
- Wauthy P, Seghers V, Mathonet P, Deuvaert FE (2009) Cardiac ochronosis: not so benign. Eur J Cardiothorac Sur 35: 732-733. [Crossref]
- Erek E, Casselman FP, Vanermen H (2004) Cardiac ochronosis: valvular heart disease with dark green discoloration of the leaflets. Tex Heart Inst J 31: 445-447. [Crossref]
- Groseanu L, Marinescu R, Laptoiun D, Botezatu I, Staniceanu F, et al. (2010) A late and difficult diagnosis of ochronosis. J Med Life 3: 437-443. [Crossref]
- Cobos Soler FJ, Molero Cabrilla R (2002) Ochronosis: a case report with multisystemic affectation, including pericardium. An Med Interna 19: 583-585. [Crossref]
- Gilbert-Barness E (2004) Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 34: 15-34. [Crossref]
- Ptacin M, Sebastian J, Bamrah VS (1985) Ochronotic cardiovascular disease. Clin Cardiol 8: 441-445. [Crossref]
- Otto C, Lind B, Kitzman D, Gersh B, Siscovick D (1999) Association of Aortic-Valve Sclerosis with Cardiovascular Mortality and Morbidity in the Elderly. N Engl J Med 341: 142-147. [Crossref]
- Hannoush H, Introne WJ, Chen MY, Lee SJ, O’Brien K, et al. (2012) Aortic stenosis and vascular calcifications in alkaptonuria. Mol Genet Metab 105: 198-202. [Crossref]
- Gaines JJ Jr, Pai GM (1987) Cardiovascular Ochronosis. Arch Pathol Lab Med 111: 991-994. [Crossref]
- Helliwell T, Gallagher J, Ranganath L (2008) Alkaptonuria - a review of surgical and autopsy pathology. Histopathology 53: 503-512. [Crossref]
- Introne WJ, Perry MB, Troendle J, Tsilou E, Kayser MA, et al. (2011) A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab 103: 307-314. [Crossref]
- Goodfellow R, Schwartz J, Leya F (2005) Black aorta: a rare finding at aortic valve replacement. J Invasive Cardiol 17: 165-167. [Crossref]
- Mayatepek E, Kallas K, Anninos A, Müller E (1998) Effects of ascorbic acid and low- protein diet in alkaptonuria. Eur J Pediatr 157: 867-868. [Crossref]
- Maceira AM, Prasad SK, Hawkins PN, Roughton M, Pennell DJ (2008) Cardiovascular magnetic resonance and prognosis in cardiac amyloidosis. J Cardiovasc Magn Reson 10: 54. [Crossref]
- Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, et al. (2008) Cardiovascular Magnetic Resonance in Clinically Suspected Cardiac Amyloidosis. J Am Coll Cardiol. 51: 1022-1030. [Crossref]
- Austin BA, Tang WH, Rodriguez ER (2009) Delayed hyper-enhancement magnetic resonance imaging provides incremental diagnostic and prognostic utility in suspected cardiac amyloidosis. J Am Coll Cardiol Img 2: 1369-1377. [Crossref]
- Migrino RQ, Christenson R, Szabo A, Bright M, Truran S, et al. (2009) Prognostic implication of late gadolinium enhancement on cardiac MRI in light chain (AL) amyloidosis on long term follow up. BMC Med Phys 9: 5. [Crossref]
- Ruberg FL, Appelbaum E, Davidoff R (2009) Diagnostic and prognostic utility of cardiovascular magnetic resonance imaging in light-chain cardiac amyloidosis. Am J Cardiol 103: 544-549. [Crossref]
- Mekinian A, Lions C, Leleu X (2010) Prognosis assessment of cardiac involvement in systemic AL amyloidosis by magnetic resonance imaging. Am J Med 123: 864-868. [Crossref]
- Aquaro GD, Pugliese NR, Perfetto F, Cappelli F, Barison A, et al. (2014) Myocardial signal intensity decay after gadolinium injection: a fast and effective method for the diagnosis of cardiac amyloidosis. Int J Cardiovasc Imaging 30: 1105-1115. [Crossref]
- White JA, Kim HW, Shah D (2014) CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. J Am Coll Cardiol Img 7: 143-156. [Crossref]
- Fontana M, Pica S, Reant P (2015) Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 132: 1570-1579. [Crossref]
- Lin L, Li X, Feng J, Shen KN, Tian Z, et al. (2018) The prognostic value of T1 mapping and late gadolinium enhancement cardiovascular magnetic resonance imaging in patients with light chain amyloidosis. J Cardiovasc Magn Reson 20: 2. [Crossref]
- Klein AL, Hatle LK, Burstow DJ, Seward JB, Kyle RA, , et al. (1989) Doppler characterization of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 13: 1017-1026. [Crossref]
- Klein AL, Hatle LK, Burstow DJ, Taliercio CP, Seward JB, et al. (1990a) Comprehensive Doppler assessment of right ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 15: 99-108. [Crossref]
- Klein AL, Hatle LK, Taliercio CL, Tayler CL, Kyle RA, et al. (1990b) Serial Doppler echocardiographic follow-up of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 16: 1135-1141. [Crossref]
- Tei C, Dujardin KS, Hodge DO, Kyle RA, Tajik AJ, et al. (1996) Doppler index combining systolic and diastolic myocardial performance: Clinical value in cardiac amyloidosis. J Am Coll Cardiol 28: 658-664. [Crossref]
- Dubrey SW, Cha K, Skinner M, LaValley M, Falk RH (1997) Familial and primary (AL) cardiac amyloidosis: Echocardiographically similar diseases with distinctly different clinical outcomes. Heart 78: 74-82. [Crossref]
- Ng B, Connors LH, Davidoff R, Skinner M, Falk RH (2005) Senile systemic amyloidosis presenting with heart failure: A comparison with light chain-associated amyloidosis. Arch Intern Med 165: 1425-1429. [Crossref]
- Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, et al. (2013) Senile systemic amyloidosis: Clinical features at presentation and outcome. J Am Heart Assoc 2: e000098. [Crossref]
- Pagourelias ED, Mirea O, Duchenne J, Van Cleemput J, Delforge M, et al. (2017) Echo parameters for differential diagnosis in cardiac amyloidosis: a head-to-head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging 10: e005588. [Crossref]
- Zhao L, Tian Z, Fang Q (2016) Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovasc Disord 16: 129. [Crossref]
- Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, et al. (2018) Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers. BMC Cardiovasc Disord 8: 221. [Crossref]
- Koyama J, Ikeda SI, Ikeda U (2015) Echocardiographic assessment of the cardiac amyloidoses. Circ J 2015: CJ-14. [Crossref]
- Raina S, Lensing SY, Nairooz RS, Pothineni NV, Hakeem A, et al. (2016) Prognostic value of late gadolinium enhancement CMR in systemic amyloidosis. JACC: Cardiovasc Imaging 9: 1267-1277. [Crossref]
- Nakahashi T, Arita T, Yamaji K, Inoue K, Yokota T, et al. (2014) Impact of clinical and echocardiographic characteristics on occurrence of cardiac events in cardiac amyloidosis as proven by endomyocardial biopsy. Int J Cardiol. 176: 753-759. [Crossref]