Abstract
A simple, rapid and sensitive method was developed and validated using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) for determination of Bezafibrate (BEZ) in human plasma. The plasma samples were extracted by protein precipitation using bezafibrate-d4 as Internal Standard (IS). The chromatographic separation was performed on Sunfire C18, 3.5µ, 2.1x50mm with a mobile phase consisting of formic acid, water and acetonitrile at flow rate of 0.30 mL/min. BEZ was detected and identified by mass spectrometry with Electrospray Ionization (ESI) in negative ion and Single Ion Recording (SIR) mode. The method was linear in the range of 100-20000 ng/mL for BEZ. The lower limit of quantification was 100 ng/mL. This method could be applied to bioequivalence studies in human plasma samples.
Key words
bezafibrate, bezafibrate-d4, human plasma, LC-MS/MS
Introduction
Bezafibrate (2-[4-[2-(4-Chlorobenzamido)ethyl]phenoxy]-2-methylpropanoic acid) is a lipid lowering agent that lowers triglycerides, LDL-cholesterol, VLDL-cholesterol and increases HDL-cholesterol levels. The activity of triglyceride lipases (lipoprotein lipase and hepatic lipoprotein lipase) which are involved in the catabolism of triglyceride-rich lipoproteins is increased by bezafibrate. Its empirical formula is C19H20ClNO4 and molecular weight is 361.82 g/mol [1-3].
Common methods for the determination of bezafibrate in human plasma are based on HPLC techniques utilizing either Protein Precipitation (PP) or Liquid-Liquid Extraction (LLE) for sample preparation [3-5]. However, the procedures employed in these studies have relatively longer run times, complicated sample preparation steps and high volumes of extraction solvents and/or mobile phases.
In this study, a simple, rapid, sensitive and cost-efficient method was developed for the determination of bezafibrate in human plasma by liquid chromatography-tandem mass spectrometry using protein precipitation to be applied to pharmacokinetic and bioequivalence studies. The method developed was fully validated as per the US Food and Drug Administration (FDA) guidelines [6] and European Medicines Agency Guideline on Bioanalytical Method Validation [7].
Experimental
Chemicals and materials
Bezafibrate (purity 99.8%) was kindly supplied by Jiuzhou Pharmaceutical (Taizhou, China). Bezafibrate-d4 (internal standard, IS) was purchased from Clearsynth (Mumbai, India). Methanol, acetonitrile and formic acid were purchased from Merck (Darmstadt, Germany). Dipotassium ethylene diamine tetra acetic acid (K2EDTA) blank human plasma was obtained from Equitech Enterprises Inc (Texas, USA).
Stock solutions, calibration standards and QCs
Stock standard solutions of BEZ were prepared in methanol at a concentration of 1 mg/mL. Working solutions in the concentration range of 100-20000 ng/mL were prepared by diluting in methanol. The working IS was prepared in methanol at concentration of 20 g/mL. Stock solutions of BEZ and IS were stored at -20ºC. Calibration standards were prepared by spiking the appropriate amounts of standard solutions into blank plasma to obtain final concentration levels of 100, 200, 500, 2500, 5000, 10000, 18000, 20000 ng/mL. The Quality Control (QC) samples were prepared at concentrations of 100, 300, 8000, 15000 and 20000 ng /mL. Calibration standards and QC samples were stored at -freezer until analysis.
Instrumentation
The LC-MS/MS system is consisted of a Waters ACQUITY LC system and a tandem quadrupole (TQ) detector with an electrospray ionization (ESI) interface (Waters Corp., Milford, MA, USA). All data were processed by MassLynx 4.1 software with the QuanLynx program (Waters Corp., USA). Separations were carried out on Sunfire C18 (3.5 µ, 2.1×50 mm) at 40 ºC. Mobile phase was consisted of acetonitrile, water and formic acid (500:500:1 v/v/v). The chromatographic run was performed under isocratic conditions at the flow rate of 0.3 mL/min and run time was 2.5 minutes. 5 µL sample was injected into the sampling system with the autosampler conditioned at 10 ºC.
Mass spectrometric detection was performed on an Acquity Tandem Quadrupole Detector in negative mode. The Single Ion Recording (SIR) transitions were performed at m/z 360.01 for BEZ and m/z 364.01 for IS with dwell time 0.5 s. The desolvation gas and cone gas flow rates and the capillary voltage were 500 L/min, 50 L/min and 3 kV, respectively. The cone voltage was set at 30V for BEZ and IS. Source temperature and desolvation temperature was set as 150 °C and 400 , respectively. The gas used for desolvation and cone was high pure nitrogen generated by Peak Scientific NL-60. MS data acquisition was conducted with the SIR mode in order to quantify and identify the investigated analytes.
Sample Preparation
Aliquots of 100 µL plasma samples and 50 µL of IS (20 µg/mL) was added into 10 mL centrifuge tube and vortexed for 5 sec. The mixture was precipitated with 1 mL acetonitrile. After vortexing for 30 sec, the samples were centrifuged at 4600 rpm for 10 min. An aliquot of 5 µL of the supernatant was injected into the LC-MS/MS system for analysis.
Results and discussion
Method Validation
Selectivity: The selectivity of the method was assessed in 10 different batches of blank plasma. These sources were also included hemolytic and lipemic K2EDTA plasma. Peak responses in blank lots were compared against the response of spiked LLOQ and no interferences were observed at the retention times of analyte and IS. The peak area of BEZ at respective retention time in blank samples should not exceed 20% of mean peak area of LLOQ of BEZ. Similarly peak area of IS at respective retention time in blank sample should not exceed 5% of mean peak area of LLOQ of IS. The selectivity of the method was demonstrated with the chromatograms of blank plasma and LLOQ samples (Figure 1). The possible interference between the BEZ, IS, paracetamol was investigated by analysing each analyte separately in triplicate (QC High concentration for BEZ, IS sample for bezafibrate-d4 10 µg/mL paracetamol in plasma were used). These samples and LLOQ samples were extracted separately (n=3) and analysed. No interference was observed.
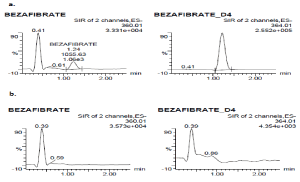
Figure 1. SIR chromatograms of 100 ng/mL (LLOQ) of BEZ spiked with internal standard (a), and human blank plasma (b)
Linearity: A calibration curve was constructed from blank sample (processed matrix sample without analyte and without IS), a zero sample (processed matrix with IS) and eight non-zero samples. Range was 100-20000 ng/mL for BEZ including the LLOQ. The linearity of each calibration curve was determined by plotting the peak-area ratio (y) of analyte to IS versus the nominal concentration (x) of analyte. Calibration curves were linear with coefficient of correlation (r2) values more than 0.9971. The r2 values, slopes and intercepts were calculated using weighted (1/X2) linear regression analysis with five intra and inter day calibration curves. The standard concentration must be within 15% deviation from the nominal value except at LLOQ, for which the maximum acceptable deviation was set as 20%. At least 75% of eight non-zero samples must be met exception criteria including acceptable LLOQ and ULOQ [6-7].
Accuracy and Precision: The within-batch precision and accuracy were determined by analyzing six replicates of quality control samples (100 ng/mL (LLOQ), 300 ng/mL (QC Low), 8000 ng/mL (QC Medium), 15000 ng/mL (QC High) and 20000 ng/mL (ULLOQ)) in a batch. The between-batch precision and accuracy was determined by analyzing six replicates of quality control samples on three different batches. The acceptance criteria of within- and between-batch weren’t exceed 15% for QC samples, expected for LLOQ which was not exceed 20%. The data on within- and between-batch precision and accuracy of the method were summarized in table 1a and table 1b.
Table 1a. Within-batch precision and accuracy of the method for determining BEZ in plasma samples
|
Batch No:1 (n=6) |
Batch No:2 (n=6) |
Batch No:3 (n=6) |
Nominal Conc.
(ng/mL) |
Conc. Found
(mean
± SD; ng/mL) |
RD
(%) |
CV (%) |
Conc. Found
(mean
± SD; ng/mL) |
RD
(%) |
CV
(%) |
Conc. Found
(mean
± SD; ng/mL) |
RD
(%) |
CV
(%) |
100 |
99.18 ± 3.63 |
-0.82 |
3.66 |
100.14 ± 1.94 |
0.15 |
1.94 |
100.71 ± 2.05 |
0.71 |
2.04 |
300 |
286.80 ± 8.22 |
-4.39 |
2.87 |
299.58 ± 3.18 |
-0.14 |
1.06 |
293.88 ± 6.92 |
-2.04 |
2.36 |
8000 |
8212.98 ± 79.89 |
2.66 |
0.97 |
8161.69 ± 74.77 |
2.02 |
0.92 |
8023.17 ± 133.53 |
-0.29 |
1.66 |
15000 |
15213.19 ± 380.77 |
1.42 |
2.50 |
14902.74 ± 219.00 |
-0.65 |
1.47 |
14957.35 ± 148.4 |
-0.28 |
0.99 |
20000 |
18775.05 ± 298.50 |
-6.13 |
1.59 |
19878.65 ± 412.28 |
-0.61 |
2.07 |
20223.16 ± 139.27 |
1.12 |
0.69 |
Table 1b. Between-batch precision and accuracy of the method for determining BEZ in plasma samples
|
Batch No:1-3 (n=18) |
Nominal Concentration (ng/mL) |
Conc. Found
(mean ± SD; ng/mL) |
RD (%) |
CV (%) |
100 |
100.01 ± 2.58 |
0.01 |
2.58 |
300 |
293.42 ± 8.12 |
-2.19 |
2.77 |
8000 |
8132.61 ± 124.79 |
1.65 |
1.53 |
15000 |
15024.43 ± 287.43 |
0.16 |
1.91 |
20000 |
19625.62 ± 697.04 |
-1.87 |
3.55 |
n: Replicates at each concentrations; RD: Relative Deviation; CV: Coefficient of Variation; SD: Standard Deviation
Matrix Effect: Matrix effect was investigated by extracting blank plasma from six different sources, including one hemolytic and one lipemic plasma. Experiments were performed at low (QC2) and high (QC4) quality control levels in six replicates using the ratios of peak areas of the blank plasma samples spiked after extraction to those of pure standard solutions containing analyte at the same concentrations. The precision (%CV) of QC2 and QC4 were 5.17% and 2.51%, respectively. The matrix effect results were summarized in table 2a and table 2b. The acceptable precision (%CV) should be ≤ 15%. According to these values, our results revealed that no significant matrix effect was observed in all these six lots of human plasma for the analyte at QC2 and QC4 concentrations.
Table 2a. Results of matrix effects for QC2 level (n=6)
Plasma QC2 |
Mean Peak Area |
Matrix Factor |
Mean Peak Area IS (n=6) |
IS Matrix Factor |
IS Normalized Matrix Factor |
Pure Solution |
2606.87 |
- |
82599.90 |
- |
- |
Matrix 1 |
3941.86 |
1.51 |
105689.18 |
1.28 |
1.18 |
Matrix 2 |
4024.05 |
1.54 |
108777.47 |
1.32 |
1.17 |
Matrix 3 |
3959.82 |
1.52 |
105321.74 |
1.28 |
1.19 |
Matrix 4 |
3897.73 |
1.50 |
103448.29 |
1.25 |
1.19 |
Matrix 5 |
3864.91 |
1.48 |
108066.50 |
1.31 |
1.13 |
Matrix 6 |
4020.49 |
1.54 |
122615.86 |
1.48 |
1.04 |
| |
|
|
|
Mean IS Normalized Matrix Factor |
1.15 |
| |
|
|
|
CV (%) |
5.17 |
|
|
|
|
|
|
Table 2b. Results of matrix effects for QC4 level (n=6)
Plasma QC4 |
Mean Peak Area |
Matrix Factor |
Mean Peak Area IS (n=6) |
IS Matrix Factor |
IS Normalized Matrix Factor |
Pure Solution |
139631.99 |
- |
82599.90 |
- |
- |
Matrix 1 |
176521.5 |
1.26 |
105689.18 |
1.280 |
0.99 |
Matrix 2 |
175555.40 |
1.26 |
108777.47 |
1.317 |
0.95 |
Matrix 3 |
172730.56 |
1.24 |
105321.74 |
1.275 |
0.97 |
Matrix 4 |
169524.34 |
1.21 |
103448.29 |
1.252 |
0.97 |
Matrix 5 |
168302.61 |
1.21 |
108066.50 |
1.308 |
0.92 |
Matrix 6 |
194735.13 |
1.40 |
122615.86 |
1.484 |
0.94 |
| |
|
|
|
Mean IS Normalized Matrix Factor |
0.96 |
| |
|
|
|
CV (%) |
2.51 |
Recovery: Recovery of BEZ was evaluated by comparison of analyte responses of six extracted samples of low, medium and high quality control concentrations (300, 8000, 15000 ng/mL) with those of six appropriately diluted standard solutions. The mean overall recovery of BEZ was 83.80±4.55%.
For IS, mean internal standard responses of six extracted samples (medium quality control concentration, 8000 ng/mL) were compared with the mean internal standard responses of six appropriately diluted internal standard solutions. Mean recovery of internal standard was 84.83%.
Stability:Stability evaluations in matrix were made using freshly spiked calibration standards. Analytes were stable up to 5 hours on bench top (mean temperature was 22 ºC) and over 3 freeze thaw cycles. The processed samples were stable up to 27 hours in autosampler at 10 ºC. Long term plasma stability was evaluated at -70 ºC and -20 ºC over a period of 8 days. The stability results were summarized in table 3.
Table 3. Results of stability of BEZ in human plasma under different storage conditions (n=6)
|
Nominal Conc.
(ng/mL) |
Conc. Found
(mean
± SD; ng/mL) |
CV (%) |
RD (%) |
Autosampler stabilitya |
300 |
296.58 ± 4.79 |
1.61 |
-1.14 |
8000 |
8039.63 ± 152.64 |
1.89 |
0.50 |
15000 |
15308.42 ± 337.24 |
2.20 |
2.06 |
|
|
|
|
|
Short-term plasma stabilityb |
300 |
304.57 ± 6.30 |
2.07 |
1.52 |
8000 |
8294.52 ± 76.20 |
0.92 |
3.68 |
15000 |
15384.71 ± 208.51 |
1.36 |
2.57 |
|
|
|
|
|
Long-term plasma stabilityc |
300 |
300.02 ± 9.33 |
3.11 |
0.01 |
8000 |
8037.43 ± 146.65 |
1.82 |
0.47 |
15000 |
14820.80 ± 508.26 |
3.43 |
-1.20 |
|
|
|
|
|
Long-term Plasma stabilityd |
300 |
302.23 ± 5.62 |
1.86 |
0.74 |
8000 |
8233.81 ± 223.99 |
2.72 |
2.92 |
15000 |
15290.59 ± 322.82 |
2.11 |
1.94 |
|
|
|
|
|
Freeze-thaw stabilityd |
300 |
293.46 ± 8.21 |
2.80 |
-2.18 |
8000 |
8018.87 ± 169.12 |
2.11 |
0.24 |
15000 |
15275.79 ± 329.12 |
2.15 |
1.84 |
RD: Relative Deviation (Accuracy), CV: Coefficient of Variation (Precision), SD: Standard Deviation
aKept at autosampler temperature, 10 0C.
bStored at room temperature.
cStored at -20 0C.
dStored at -70 0C.
Conclusion
A simple, fast, accurate and sensitive LC-MS/MS method using SIR ion mode was developed for the analysis of BEZ in human plasma utilizing a simple extraction procedure and short run time which are advantageous to be employed for batches with large number of samples. The method was fully validated in accordance to FDA and EMA guidelines. The results of the validation studies demonstrated that the developed method had high sensitivity, recovery, precision, accuracy and reproducibility. Finally, the method could be applied to bioequivalence studies to evaluate the pharmacokinetics of BEZ.
References
- Ledermann H, Kaufmann B (1981) Comparative pharmacokinetics of 400 mg bezafibrate after a single oral administration of a new slow-release preparation and the currently available commercial form. J Int Med Res 9: 516-520. [Crossref]
- Sigma-Aldrich. Bezafibrate Material Safety Data Sheet, 2017.
- Wei Z, Bing-ren X, Ying Z, Liyan Y, Teng W, et al. (2008) J Chromatogr Sci 46: 844-847. [Crossref]
- Masnatta LD, Cuniberti LA, Rey RH, Werba JP (1996) Determination of bezafibrate, ciprofibrate and fenofibric acid in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl 687: 437-442. [Crossref]
- Castoldi D, Monzani V, Tofanetti O (1985) Determination of bezafibrate in human plasma and urine by high-performance liquid chromatography. J Chromatogr 344: 259-265. [Crossref]
- U.S. Department of Health and Human Services, Food and Drug Administration (FDA). Guidance for Industry. Bioanalytical Method Validation. 2013.
- European Medicines Agency (EMA), Committee for Medicinal Product for Human Use (CHMP), Guideline on Bioanalytical Method Validation. 2011