An isocratic HPLC method had been developed and validated for rapid simultaneous separation and determination of the two antihypertensive drugs, losartan and spironolactone in tablet dosage forms and also in presence of human plasma within 6 minutes. Separation was carried out on a Thermo Scientific® BDS Hypersil C8 column (5 µm, 2.50×4.60 mm) using a mobile phase of ACN: 0.025 M KH2PO4 (60:40, v/v) adjusted to pH 3.49 with ortho - phosphoric acid at ambient temperature. The flow rate was 1 ml/min and maximum absorption was measured using DAD detector at 235 nm. The retention times of losartan and spironolactone were recorded to be 3.47, and 4.63 minutes respectively, indicating a shorter analysis time. Limits of detection were reported to be 0.07 and 0.15 µg/ml for losartan and spironolactone, respectively, showing a high degree of the method sensitivity. The method was then validated according to FDA guidelines for the determination of the drugs either in tablets with highly precise recoveries or clinically in human plasma specially regarding pharmacokinetic and bioequivalence studies.
HPLC, losartan, spironolactone, tablet, human plasma
Losartan Potassium (Figure 1 LOS), is chemically 2-n-butyl-4-chloro-1-[p-(o-1Htetrazol- 5-ylphenyl) benzyl]-imidazole-5-methanol monopotassium salt. It is a highly selective, orally active, non-peptide angiotensin II receptor antagonist indicated for the treatment of hypertension [1].
Figure 1. Chemical structures of losartan (LOS) and spironolactone (SPR)
Literature survey demonstrated that few analytical techniques have been employed for the determination of losartan such as HPLC, capillary electrophoresis and super-critical fluid chromatography [1-5], conductometry [6] and spectrophotometry [7-9].
Spironolactone (Figure 1 SPR), is chemically 7α-acetylthio-3-oxo-17α-pregn-4-ene-21,17-carbolactone. Spironolactone is a selective and competitive anatagonsit of aldosterone at the intracellular aldosterone receptors in distal tubule cells. This anatagonism effect increases the excretion of water and sodium, while decreasing the excretion of potassium (K+ sparing diuretic). It has a fairly slow onset of action, taking several days to develop and similarly, the effect diminishes slowly [10].
Based on our continuous survery, we revealed that the determination of spironolactone has been carried out by HPLC in tablets [11], in plasma samples [12-15] or in both matrices [16], and also by spectrophotometry [17-19].
To the best of our knowledge and comprehensive survey, losartan and spironolactone mixture was not determined before by chromatographic techniques neither in pharmaceutical nor in biological samples despite their synergistic action. As such, the present work introduces a simple, rapid, reproducible and sensitive chromatographic method for the determination of losartan and spironolactone in both tablets and human plasma samples. The method was finally compared statistically with a reference method showing equal accuracy, reproducibility and no significant difference with the reported one.
Apparatus
- Agilent 1200® HPLC instrument (Germany) with a Thermo Scientific® BDS Hypersil C8 column (5 µm, 2.50×4.60 mm), DAD absorbance detector, HPLC QUAT pumps and connected to PC computer loaded with Agilent 1200 software.
- Labomed® Spectro UV-VIS Double Beam (UVD-2950) Spectrophotometer with matched 1 cm quartz cells and connected to windows compatible computer using UV Win 5 Software v6.
- HANNA® HI 8314 membrane pH-meter (Romania) for pH adjustment.
Materials and reagents
- All solvents and reagents were of an HPLC analytical grade (acetonitrile, potassium dihydrogen phosphate and ortho-phosphoric acid were supported from Fisher Scientific, England).
- Losartan & spironolactone were kindly provided by Kahira Pharmaceuticals & Chemicals and Amriya Pharmaceuticals, Egypt, respectively. Standard solutions of 200 µg/ml were prepared by dissolving 20 mg of each pure drug in 100 ml of the mobile phase.
- Mobile phase was a freshly prepared binary mixture of ACN: 0.025 M potassium dihydrogen phosphate (60:40, v/v) adjusted to pH 3.49 using ortho-phosphoric acid, filtered and degassed using 0.45 µm membrane filter (Millipore, USA).
- Losartan® tablets (Amriya Pharm, Egypt) and Aldactone® tablets (Kahira Pharm & Chem. Egypt) were labeled to contain 50 mg losartan and 25 mg spironolactone, respectively.
- The human plasma was kindly provided by Zagazig University Hospital and was tested to be drug and disease free. Plasma was kept frozen at -20ºC before use, and was then stored at -4ºC between uses.
Procedures
Preparation of standard calibration curves: Appropriate mixed dilutions of losartan and spironolactone standard stock solutions were done in 10 ml volumetric flasks to get final concentrations of 2.50, 5, 10, 12.50, 25, 50 and 100 µg/ml for both drugs. A 10 μl of each mixture was injected into the column and the chromatogram was obtained at 235 nm. A graph was plotted as concentration of drugs against response (peak area). Regarding validation QC samples, concentrations of 2.50, 12.50 and 50 µg/ml were selected as low (LQC), medium (MQC) and high (HQC) levels, respectively.
Pharmaceutical tablets procedure: 5 tablets of losartan® and aldactone® tablet formulations were weighed and powdered. An accurately amounts of the powder equivalent to 20 mg of each drug were dissolved in the mobile phase, filtered into 100 ml measuring flasks and completed to volume with the mobile phase. The procedure was then completed as mentioned above under the general procedure 2.3.1 applying standard addition techniques.
Human plasma samples procedure: Calibration curve and validation QC samples at concentrations of 2.50, 5 and 10 µg/ml in plasma were prepared. Aliquots of 200 µl plasma samples and different drug mixture volumes ranging from 100 up to 200 µl were added into 10 ml centrifuge tubes and vortexed for 1 min. The mixture was then precipitated with methanol (total volume 2 ml). After vortexing for 1 min, the samples were centrifuged at 5000 rpm for 15 min. A aliquots of 10 µl of the supernatant was filtered through 0.45 µm PTFE syring filters (Membrane solutions, USA) and injected into the HPLC system for analysis.
Optimization of chromatographic conditions
All chromatographic conditions are illustrated in Table 1. Spectroscopic analysis of the drugs in the range of 200-400 nm showed that losartan and spironolactone have UV absorbance maxima (λmax) at 225 nm and 238 nm, respectively as depicted in Figure 2. Therefore, the chromatographic detection was performed at 235 nm as the appropriate wavelength using a DAD detector. The method was performed on a Thermo Scientific® BDS Hypersil C8 column (5 µm, 2.50×4.60 mm).
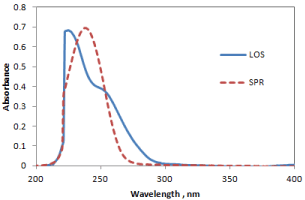
Figure 2. Overlain spectra of losartan (LOS___) and spironolactone (SPR----) at 50 µg/ml concentration
Table 1. Chromatographic conditions for the proposed method
|
Parameters
|
Conditions
|
|
Column
|
Thermo Scientific® BDS Hypersil C8 5µm (2.50×4.60 mm)
|
|
Mobile phase
|
Isocratic binary mobile phase of ACN: 0.025 M KH2PO4 adjusted to pH 3.49 using ortho - phosphoric acid (60:40, v/v), filtered and degassed using 0.45µm membrane filter.
|
|
UV detection, nm
|
235
|
|
Flow rate, ml/min
|
1
|
|
Injected volume, µl
|
10
|
|
Pressure, psig
|
98
|
|
Temperature
|
Ambient
|
Furthermore, under several trials of mobile phase optimization regarding its composition ratio and pH, it was observed that the optimized mobile phase was determined as a mixture of ACN: 0.025 M potassium dihydrogen phosphate adjusted to pH 3.49 using ortho-phosphoric acid (60:40, v/v) at a flow rate of 1 ml/min. Under these conditions, losartan and spironolactone in both pure form and tablet formulations can be separated and eluted at 3.47 and 4.63 minutes respectively as illustrated in Figures 3A and 3B, respectively. Also, the mixture determination in plasma didn't show the matrix interference effect as the human plasma was eluted at 2.55 minute in correspondence with the migration times of losartan and spironolactone (Figures 3C and 3D). However, in all cases, the optimum mobile phase showed symmetrical peaks (0.8<T<1.2), capacity factor (1<k<10), resolution>2 and theoretical plates>2000. Table 2 shows all system suitability parameters of the proposed HPLC-DAD method for simultaneous determination of the two antihypertensive drugs in both pure and plasma samples.
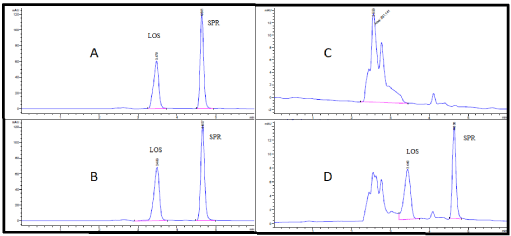
Figure 3. HPLC Chromatogram of (A) authentic mixture of losartan and spironolactone, (B) losartan® and Aldactone® in tablet formulation mixture, (C) blank human plasma sample, and (D) mixture of losartan and spironolactone in human plasma sample, all performed at the optimum conditions stated in Table 1
Table 2. System suitability parameters for losartan and spironolactone in both pure and plasma samples.
|
Parameters
|
Pure sample
|
Plasma sample
|
Reference values [23]
|
|
LOS
|
SPR
|
LOS
|
SPR
|
|
|
Retention time, tr
|
3.47
|
4.63
|
3.46
|
4.62
|
|
|
Capacity factor, k'
|
1.67
|
2.57
|
1.66
|
2.56
|
Accepted k' value (1-10)
|
|
Peak asymmetry (Tailing factor, T)
|
1.19
|
0.90
|
0.99
|
0.83
|
Accepted T value ≤ 2
|
|
Therotical plates, N
|
3334
|
14003
|
3417
|
13315
|
Accepted N value>2000
|
|
Resolution, Rs
|
5.83
|
5.11
|
Accepted value>2
|
|
Selectivity (Separation factor, α)
|
1.53
|
1.54
|
|
Method validation
The method validation was performed according to food and drug administration and international conference of harmonization guidelines (ICH) [20-22].
Linearity: Five different concentrations of the drug mixture were specified for linearity studies. The calibration curves obtained by plotting peak area against concentration showed linearity in the concentration range of 2.50-100 µg/ml for both drugs (Table 3). Linear regression equations of losartan and spironolactone were found to be y = 21.30x +31.62 and y = 27.51x + 45.51, respectively and the regression coefficient values (r) were calculated to be 1 and 0.999, respectively indicating a high degree of linearity for both drugs (Figure S1).
Table 3. Analytical merits for determination of losartan and spironolactone in pure samples using the proposed method
|
|
LOS
|
SPR
|
|
Conc. taken (µg/ml)
|
Conc. found (µg/ml)
|
Recovery
|
% Accuracy
|
Conc. taken (µg/ml)
|
Conc. found (µg/ml)
|
Recovery
|
% Accuracy
|
|
|
2.50
|
2.51
|
100.24
|
0.24
|
2.50
|
2.49
|
101.03
|
2.75
|
|
|
12.50
|
12.36
|
98.92
|
-1.07
|
12.50
|
12.31
|
98.71
|
-1.48
|
|
|
25
|
25.04
|
100.16
|
0.16
|
25
|
24.67
|
98.78
|
-1.29
|
|
|
50
|
50.15
|
100.31
|
0.31
|
50
|
50.67
|
101.36
|
1.35
|
|
|
100
|
99.92
|
99.92
|
-0.07
|
100
|
99.72
|
99.75
|
-0.27
|
|
Mean
|
|
|
99.91
|
-0.08
|
|
|
99.93
|
0.211
|
|
SD
|
|
|
0.57
|
|
|
|
1.23
|
|
|
CV (%)
|
|
|
0.58
|
|
|
|
1.24
|
|
|
SE
|
|
|
0.25
|
|
|
|
0.55
|
|
|
Variance
|
|
|
0.33
|
|
|
|
1.52
|
|
|
Slope
|
|
|
21.29
|
|
|
|
27.51
|
|
|
LOD
|
|
|
0.07
|
|
|
|
0.15
|
|
|
LOQ
|
|
|
0.25
|
|
|
|
0.51
|
|
Accuracy: The accuracy of the method was determined by investigating the recoveries of commercial losartan and spironolactone at concentration levels covering the specified range (three replicates of each concentration) using the standard addition technique. It was performed by adding a fixed standard concentration for each drug at different levels and the proposed method was followed. From the amount of the drug estimated, the percentage recovery was calculated and the results shown in Table S1 are indicating excellent recoveries for both drugs.
Precision: The precision of the method was evaluated according to intra-day and inter-day precision using validation QC samples at concentrations of 2.50, 12.50 and 50 µg/ml. Intra-day precision was evaluated in respect of both standard deviation (SD) and coefficient of variation (CV%) regarding three replicate determinations using the same solution containing pure drugs. The SD and CV% values in Table 4 (varied from 0.04 to 1.80) revealed the high precision of the method. For inter-day reproducibility, the day-to-day SD and CV% values were also in the acceptable range of 0.15-2.79 (Table 4). These results show that the proposed method has an adequate precision in simultaneous determination of both drugs in pharmaceutical formulations.
Table 4. Intra- and inter-day precision and stability results of losartan and spironolactone in both pure and plasma samples
|
|
Drugs
|
Concentrations
(µg/ml)
|
Mean±SD
|
CV (%)
|
|
Intra-day runs
(n=3)
|
LOS
|
50
12.50
2.50
|
99.6±0.12
98.52±0.13
100.24±0.79
|
0.11
0.12
0.78
|
|
|
SPR
|
50
12.50
2.50
|
101.18±0.05
98.03±0.18
101.12±1.79
|
0.04
0.17
1.80
|
|
Inter-day runs
(n=3)
|
LOS
|
50
12.50
2.50
|
100.87±0.53
98.07±0.39
100.71±2.79
|
0.52
0.4
2.77
|
|
|
SPR
|
50
12.50
2.50
|
101.09±0.15
99.1±0.38
100.21±1.51
|
0.16
0.39
1.53
|
|
3 Freeze–thaw cycles at −20°C (n plasma=3)
|
LOS
|
10
5
2.5
|
81.08± 0.33
82.19± 1.11
81.46± 2.65
|
0.4
1.35
3.25
|
|
|
SPR
|
10
5
2.5
|
86.61±0.09
87.22±1.54
85.04±2.05
|
0.1
1.76
2.37
|
Selectivity and specificity: Selectivity of the method was checked by injecting the solutions of losartan and spironolactone into the column separately where two sharp peaks were obtained at retention times of 3.47, and 4.63 min, respectively, and these peaks were not obtained for the blank solution.
Also, the specificity studies revealed that the presence of the excipents in the tablet formulations didn't show any kind of impurity interference with the sharp and well-resolved peaks of losartan and spironolactone (Figure 3 B).
Limits of detection and limits of quantification: For determining the limits of detection and quantitation, the method based on signal-to-noise ratio (3:1 for LOD & 10:1 for LOQ) was adopted. Limits of detection were reported to be 0.07 and 0.15 µg/ml, while limits of quantification were calculated to be 0.25 and 0.51 for both losartan and spironolactone, respectively (Table 3) showing that the proposed method is highly sensitive and applicable for pharmacokinetic and bioequivalence studies where detection of small concentrations in plasma is required.
Robustness: The robustness of the methods was evaluated by making deliberate subtle changes (±0.05) in the flow rate, pH of mobile phase and mobile phase composition ratio keeping the other chromatographic conditions constant. The changes effect was studied on the basis of percent recovery and standard deviation of both drugs. Table 5 shows that the changes had negligible influences on the results as revealed by small SD values (≤1.87).
Table 5. Results of the robustness for the determination of losartan, and spironolactone (12.5 µg/ml) using the proposed method
|
Parameter
|
LOS
|
SPR
|
|
Mean recovery±SD
|
CV (%)
|
% Accuracy
|
Mean recovery±SD
|
CV (%)
|
% Accuracy
|
|
Flow rate 0.95 ml (- 0.05)
|
100.40±0.63
|
0.40
|
0.44
|
100.80±1.57
|
2.49
|
0.80
|
|
Flow rate 1.05 ml (+ 0.05)
|
100.20±0.15
|
0.02
|
0.15
|
100.56±1.54
|
2.40
|
0.57
|
|
Buffer pH 3.44 (- 0.05)
|
99.77±0.88
|
0.78
|
-0.22
|
100.16±1.87
|
3.52
|
0.17
|
|
Buffer pH 3.54 (+ 0.05)
|
100.00±0.30
|
0.09
|
0.04
|
100.28±1.74
|
3.03
|
0.28
|
|
ACN:Buffer ratio 60.5:39.5
|
100.00±0.35
|
0.12
|
0.02
|
100.42±1.61
|
2.62
|
0.42
|
|
ACN:Buffer ratio 59.5:40.5
|
100.10±0.20
|
0.04
|
0.10
|
100.31±1.70
|
2.90
|
0.32
|
Applications
Analysisof tablet formulations: Losartan® and Aldactone® pharmaceutical formulation containing losartan and spironolactone had been successfully analyzed by the proposed method. Excipients and impurities did not show interference indicating high specificity of the method. Results obtained were compared to those obtained by applying reference methods [9,11] where Student’s t-test and F-test were performed for comparison. Results shown in Table 6 indicated that calculated t and F values were less than tabulated ones for losartan and spironolactone which in turn indicate that there is no significant difference between proposed method and reference ones relative to precision and accuracy.
Table 6. Statistical analysis of results obtained by the proposed method applied on Losartan® and Aldactone® tablets compared with reference methods
|
|
LOS (Losartan®)
|
SPR (Aldactone®)
|
|
Proposed method
|
Reference
method [9]
|
Proposed method
|
Reference
method [10]
|
|
N
|
4
|
4
|
4
|
4
|
|
Mean Recovery
|
99.38
|
99.81
|
100.25
|
99.27
|
|
SE
|
0.41
|
0.16
|
0.39
|
0.68
|
|
Variance
|
0.85
|
0.11
|
0.79
|
1.85
|
|
Student-t
|
0.87 (1.94)a
|
|
1.19 (1.94)a
|
|
|
F-test
|
7.95 (9.28)b
|
|
2.32 (9.28)b
|
|
a and b are the Theoretical Student t-values and F-ratios at p=0.05.
Analysis of human plasma: The proposed method was also applied for determination of losartan and spironolactone in human plasma samples by applying protein preciptation procedure. Retention times of losartan and spironolactone in plasma samples and the other system suitability parameters were also pretty similar to those in pure and pharmaceutical ones (Table 2).
The calibration curves in the spiked plasma were also found to be linear over the clinical range of 2.50-10 μg/ml for both drugs (Table S2). Also, the plasma chromatogram (Figures 3C and 3D) confirms the specificity of the method in clinical studies as the plasma peak (eluting at 2.55 minute) is not interfering but well separated from both peaks of losartan and spironolactone. Stability and precision studies were conducted by applying plasma freeze-thaw cycles at -200C (over three days) using validation QC samples at concentrations of 2.50, 5 and 10 µg/ml of losartan and spironolactone in plasma and results are summarized in Table 4. The recoveries for losartan and spironolactone were in the range of 81.08-82.19% and 85.04-87.22%, where coefficients of variation were 0.4-3.25% and 0.1-2.37%, respectively. In terms of sensitivity, LOD, LOQ, and even migration time, the proposed method has been shown to be superior over been previously reported methods for the analysis of losartan and spironolactone in real samples or in pharmaceutical formulations [4,9,14,16].
The presented method was developed and validated for rapid simultaneous estimation of losartan and spironolactone within 6 minutes. The results obtained indicate that the proposed method is rapid, accurate, selective, robust and reproducible. Linearity was observed over a concentration range of 2.50 to 100 μg/ml for both drugs. The method has been successfully applied for the analysis of marketed tablets Losartan® and Aldactone® in respect of quality control, where low cost and fast analysis are essential. This analytical method can be also adequate and useful for the clinical estimation of losartan and spironolactone in human plasma samples according to FDA guidelines in respect of pharmacokinetic and bioequivalence studies that would be useful in therapeutic drug monitoring.
The authors would like to thank the Bioequivalence and Accurate Measurement Unit, Zagazig University for facilitating and conducting the spectroscopic and chromatoraphic study. Also, the authors are grateful to Zagazig University Hospital for providing the drug and disease free plasma samples.
The authors declare that there is no conflict of interest in the manuscript.
This manuscript does not include any studies on human or animals.
- Obando MA, Estela JM, Cerdà V (2008) Simultaneous determination of hydrochlorothiazide and losartan potassium in tablets by high-performance low-pressure chromatography using a multi-syringe burette coupled to a monolithic column. Anal Bioanal Chem 391: 2349-2356.
- Furtek CI, Lo MW (1992) Simultaneous determination of a novel angiotensin II receptor blocking agent, losartan, and its metabolite in human plasma and urine by high-performance liquid chromatography. J Chromatograph B: Biomed Sci Ap 573: 295-301.
- Williams RC, Alasandro MS, Fasone VL, Boucher RJ, Edwards JF (1996) Comparison of liquid chromatography, capillary electrophoresis and super-critical fluid chromatography in the determination of losartan potassium drug substance in Cozaar® tablets. J Pharm Biomed Anal 14: 1539-1546.
- Argekar AP, Sawant JG (2000) A gradient reversed phase high performance liquid chromatography method for simultaneous determination of hydrochlorothiazide (HCT) and losartan potassium (LOS) from tablets. Anal Lett 33: 869-880
- Anderson JF, Gerlin MG, Sversut RA, Oliveira LS, Singh AK, et al. (2017) Development and validation of an isocratic HPLC method for simultaneous determination of quaternary mixtures of antihypertensive drugs in pharmaceutical formulations. Acta Chromatogr 29: 95-110.
- Oliveira RP, Felix F, Angnes L (2012) A simple and precise conductometric method for the determination of losartan in pharmaceutical products. Open Chem 10: 1842-1849.
- Lastra OC, Lemus IG, Sánchez HJ, Pérez RF (2003) Development and validation of an UV derivative spectrophotometric determination of losartan potassium in tablets. J Pharm.Biomed Anal 33: 175-180.
- Nagavalli D, Vaidhyalingam V, Santha A, Sankar AS, Divya O (2010) Simultaneous spectrophotometric determination of losartan potassium, amlodipine besilate and hydrochlorothiazide in pharmaceuticals by chemometric methods. Acta Pharm 60: 141-152.
- Wankhede SB, Raka KC, Wadkar SB, Chitlange SS (2010) Spectrophotometric and HPLC methods for simultaneous estimation of amlodipine besilate, losartan potassium and hydrochlorothiazide in tablets. Ind J Pharm Sci 72: 136-140.
- Ram VR, Dave PN, Joshi HS (2012) Development and validation of a stability-indicating HPLC assay method for simultaneous determination of spironolactone and furosemide in tablet formulation. J Chromatogr Sci 50: 721-726.
- Vijay RR, Pragnesh ND, Hitendra SJ (2012) Development and validation of a stability-indicating HPLC assay method for simultaneous determination of spironolactone and furosemide in tablet formulation. J Chromatogr Sci 50: 721-726.
- Dong H, Xu F, Zhang Z, Tian Y, Chen Y (2006) Simultaneous determination of spironolactone and its active metabolite canrenone in human plasma by HPLC‐APCI‐MS. J Mass Spect 41: 477-486.
- Sandall JM, Millership JS, Collier PS, McElnay JC (2006) Development and validation of an HPLC method for the determination of spironolactone and its metabolites in paediatric plasma samples. J Chromatogr B 839: 36-44.
- Sultana N, Arayne MS, Iftikhar B (2008) Simultaneous determination of atenolol, rosuvastatin, spironolactone, glibenclamide and naproxen sodium in pharmaceutical formulations and human plasma by RP‐HPLC. J Chin Chem Soc 55: 1022-1029.
- Vlase L, Imre S, Muntean D, Achim M, Muntean DL (2011) Determination of spironolactone and canrenone in human plasma by high-performance liquid chromatography with mass spectrometry detection. Cro Chem Acta 84: 361-366.
- Walash MI, El-Enany N, Eid MI, Fathy ME (2012) Micellar high performance liquid chromatographic determination of furosemide and spironolactone in combined dosage forms. Application to human plasma. J Pharm Res 5: 2648-2656.
- Martı́n E, Jimenez AI, Hernandez O, Jiménez F, Arias JJ (1999) Simultaneous kinetic spectrophotometric determination of spironolactone and canrenone in urine using partial least-squares regression. Talanta 49: 143-154.
- Dinç E, Üstündağ Ö (2003) Spectophotometric quantitative resolution of hydrochlorothiazide and spironolactone in tablets by chemometric analysis methods. II Farmaco 58: 1151-1161.
- Millership JS, Parker C, Donnelly D (2005) Ratio spectra derivative spectrophotometry for the determination of furosemide and spironolactone in a capsule formulation. II Farmaco 60: 333-338.
- Guidance for Industry: Q2B validation of analytical procedures: Methodology. International conference of harmonization (ICH). Nov. 1996. (https://www.fda.gov/downloads/drugs/guidances/ucm073384.pdf).
- US Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation, 2001. (http://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf).
- Zimmer D (2014) New US FDA draft guidance on bioanalytical method validation versus current FDA and EMA Guidelines: chromatographic methods and ISR. Bioanal 6: 13-19.
- CDER Center for Drug Evaluation and Research, Reviewer Guidance Validation of Chromatographic Methods, 1994. (https://www.fda.gov/downloads/drugs/guidances/ucm134409.pdf).