Abstract
Background: There is growing evidence that diabetes mellitus modifies the pharmacokinetics of several medications, changing their pharmacodynamics. To understand these changes, in this study, we develop a physiologically based pharmacokinetic (PBPK) model to predict drug disposition in diabetic patients.
Methods: The PBPK model coupled with the GastroPlusTM version 9.5 software (Simulation Plus Inc., Lancaster, CA) was utilized to predict the means and variability of pharmacokinetic parameters. A whole-body PBPK model with key diabetic-related physiological changes was developed, in order to characterize the pharmacokinetics of the tested drugs in diabetic patients and compare these parameters to those in healthy subjects. Data related to physiological and biological changes in diabetic patients were obtained from the literature and incorporated into a structural PBPK model describing the PK data of healthy subjects.
Results: Glibenclamide (mainly metabolized by CYP2C9) and chlorzoxazone (metabolized by CYP2E1 and CYP1A2) were used for model development and validation. Changes in glibenclamide and chlorzoxazone area under the curve (AUC) and maximum concentration (Cmax) values in diabetic patients were predicted. The simulated PK profiles were comparable with the observed values, with predicted-to-observed ratio in the range of 0.8–1.2.
Conclusion: In vitro data and information from a healthy population were successfully used to predict the pharmacokinetic profiles of medications in diabetic patients using the developed DM-PBPK model, which included changes in numerous anatomical, physiological, and biochemical characteristics caused by diabetes. When no clinical trials are available to guide dose recommendations in diabetic patients, the DM-PBPK model offers a workable substitute for empirical dosage selection.
Key words
Diabetes mellitus, pharmacokinetics, PBPK, glibenclamide, chlorzoxazone
Introduction
Diabetes Mellitus (DM) is a growing global health concern. It is a metabolic disease of multiple etiologies, characterized by elevated blood sugar resulting from defects in insulin secretion, insulin action, or both [1]. In addition, DM is associated with carbohydrate, fat, and protein metabolism disturbances. Patients with long-term diabetes usually experience serious macro- and micro-vascular complications including cardiovascular and renal disease [1].
There have been a limited number of studies related to the influence of DM on drug pharmacokinetics. This limited information translates to an absence of available guidelines that help healthcare providers, in terms of adjusting dosages for diabetic patients to reach an appropriate therapeutic effect. Pathophysiological changes during diabetes have the potential to affect the absorption, distribution, metabolism, and excretion (ADME) of various medications [2].
Considering absorption, studies have shown that an increase in extracellular glucose can affect membrane permeability, thus affecting the absorption of various medications (i.e., bioavailability) [2,3]. Moreover, it has been reported that 28‒65% of diabetic patients experience delayed gastric emptying, which may have a significant impact on the absorption of oral medications [2,3]. In the context of distribution, DM can affect the distribution of medications by affecting blood proteins [4,5]. Albumin is a major protein in blood that binds with medications [4,5]. It can become glycated in the presence of high glucose concentrations [4,5], leading to a conformational change in the albumin structure that alters the fraction of unbound drugs [4,5]. Moreover, increased levels of free fatty acids in diabetes patients may decrease the ability of drugs to bind to albumin [2]. In terms of metabolism, it has been stated that diabetes is associated with a significant change in Cytochrome (CYP3A4), which is responsible for the metabolism of medications in the liver [2,6].
Regarding excretion, it is well-known that long-term uncontrolled diabetes can lead to an increased glomerular filtration rate (GFR) [2]. A report has shown that the plasma concentrations of drugs predominantly excreted by the kidney were negatively correlated with GFR [7]. Furthermore, 40% of DM patients were suggested to develop nephropathy, also affecting the clearance of medications [8]. Obesity is often associated with type 2 diabetes mellitus, leading to metabolic abnormalities under diabetic conditions [9]. Moreover, the changes in tissue blood flow induced by diabetes have an impact on the pharmacokinetic behaviors of drugs [7].
All of these changes could lead to significant alterations to the pharmacokinetic parameters of drugs, especially narrow therapeutic drugs. Therefore, it is necessary to predict pharmacokinetic behaviors accurately in diabetic patients, in order to optimize the dose appropriately. Accordingly, we decided to build a Physiologically Based Pharmacokinetic (PBPK) model that can predict the alteration of pharmacokinetics in diabetic patients using data available in the literature.
The PBPK model incorporates a wide range of physiological and biochemical parameters that are altered under the diabetic condition, including the gastric emptying rate, intestinal transit time, drug metabolism capacity with respect to liver and kidney functions, tissue volume, and blood flow. This allows for extrapolation of the in vivo pharmacokinetics of drugs in diabetic populations [2].
Glibenclamide (mainly metabolized by CYP2C9) and chlorzoxazone (metabolized by CYP2E1 and CYP1A2) were used for model development and validation. For chlorzoxazone, virtual trials using the GastroPlusTM version 9.5 software, embedded with the Advanced Compartmental Absorption Transit (ACAT) model and PBPKPlus™ module, were used for analysis of the inter-subject variability of the model and for internal model validation. The plasma concentration–time profiles of drugs in healthy subjects and diabetic patients were simultaneously simulated and compared with clinical reports.
Methods
General workflow of PBPK model development and validation process: All the PBPK simulations were carried out using the commercially available software GastroPlusTM version 9.5 (Simulation Plus Inc., Lancaster, CA), embedded with the Advanced Compartmental Absorption Transit (ACAT) model and PBPKPlus™ module. The general workflow of PBPK modeling and simulation of test compounds in non-diabetic and diabetic subjects are presented in Figure 1.
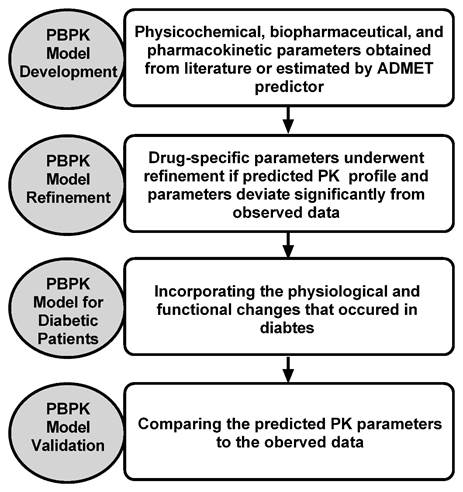
Figure 1. Diagram representing the general workflow of the PBPK model development and verification for the test compounds
Glibenclamide PBPK model development: The glibenclamide PBPK model was initially developed using physicochemical, biopharmaceutical, and PK parameters obtained from the literature or estimated using the ADMET Predictor (summarized in Table 1). Absorption was predicted by the ACAT model, which was used as an input into the PBPK model, in order to predict the plasma and tissue concentration–time profiles after oral administration. We assumed that glibenclamide reaches rapid partitioning equilibrium between homogenous tissues and plasma/blood and, thus, perfusion-limited distribution kinetics was considered for all tissues in the PBPK model. The steady-state volume of distribution (Vss) and tissue–plasma partition coefficients (Kp) were predicted using the algorithms of Rodgers et al. [10].
Table 1. Physicochemical and in vitro data used in the glibenclamide PBPK model
Parameter |
Value |
references |
Molecular weight |
494 g/mol |
- |
Log P |
4.5 |
[19] |
pKa |
5.3 |
[19, 20] |
Solubility |
0.08 mg/ml |
ADMET Predictor |
Effective permeability |
5.6 × 10-4 cm/s |
[19] |
Fraction unbound |
0.01 |
[21-23] |
Blood:Plasma ratio |
0.55 |
[24] |
Vd (L/kg) |
0.125 L/kg |
[25] |
CLR (L/h) |
0 L/h |
[21] |
|
Metabolism |
Vmax (pmol/min/pmol of isoform) |
Km (μM) |
|
CYP3A4 |
14.4 |
5.2 |
[11] |
CYP3A5 |
1.9 |
4.2 |
[11] |
CYP2C19 |
9.6 |
15.1 |
[11] |
CYP2C8 |
2.5 |
7.7 |
[11] |
CYP2C9 |
1.0 |
4.7 |
[11] |
Glibenclamide is mainly cleared through metabolism by the CYP system, primarily by CYP3A family enzymes [11]. CYP2C19, CYP28, and CYP2C9 contribute to the metabolism of glibenclamide, to a lesser extent [11]. The hepatic intrinsic metabolic clearance (CLint,H) of glibenclamide was predicted using in vitro to in vivo extrapolation from in vitro studies [11]. The contribution of intestinal metabolism was also considered in the simulation, using the in vitro KM and Vmax values for CYP3A4, CYP3A5, CYP2C19, CYP28, and CYP2C9.
The initial model was verified by comparing the simulated PK profiles with the observed data from several single and multiple dosing studies in healthy subjects and patients [12,13]. If the predicted PK profile and parameters significantly deviated from the observed data, the model would then be refined by parameter optimization by fitting against the observed clinical data. The GastroPlus optimization module was utilized to estimate the parameters that significantly influence PK profiles. The estimated Kp values were optimized to fit the plasma profiles reported in clinical trials. Initial scaling using enzyme kinetics values for both enzymes (determined in vitro) significantly under-predicted in vivo IV clearance (CLIV) and CLORAL. The enzyme kinetics parameters were, therefore, optimized using the GastroPlus build-in optimization moduleTM, in order to capture the PK profile of glibenclamide in healthy volunteer clinical studies. The verified PBPK model was then used to evaluate the effect of diabetes mellitus on the dynamics of glibenclamide.
Chlorzoxazone PBPK model development: Similarly, the chlorzoxazone PBPK model was initially developed using physicochemical, biopharmaceutical, and PK parameters obtained from the literature or estimated by the ADMET Predictor (summarized in Table 2). Perfusion-limited distribution kinetics were considered for all tissues in the PBPK model. Vss and Kp were predicted using the algorithms of Rodgers et al. [14].
Table 2. Physicochemical and in vitro data used in the chlorzoxazone PBPK model
Parameter |
Value |
references |
Molecular weight |
169.5 g/mol |
- |
Log P |
1.94 |
ADMET Predictor |
pKa |
9.3 |
ADMET Predictor |
Solubility |
2.96 |
ADMET Predictor |
Effective permeability |
6.5 × 10-4 cm/s |
[26] |
Fraction unbound |
0.05 |
PBPK Plus |
Blood:Plasma ratio |
0.05 |
PBPK Plus |
Vd (L/kg) |
0.125 L/kg |
[17] |
CLR (L/h) |
0 L/h |
[16] |
|
Metabolism |
Vmax (nmol/min/mg) |
Km (μM) |
|
CYP2E1 |
1.6 |
70.5 |
[15] |
Chlorzoxazone is extensively metabolized after administration [15]. CYP2E1 is the enzyme responsible for the metabolism of chlorzoxazone [15,16]. The hepatic intrinsic metabolic clearance (CLint,H) of chlorzoxazone was predicted using in vitro to in vivo extrapolation from in vitro studies [15]. The metabolism parameters (Vmax and Km) were taken from the literature and optimized to fit the observed data [15]. The contribution of intestinal metabolism was also considered in the simulation.
The initial model was verified by comparing the simulated PK profiles with the observed data from several single and multiple dosing studies in healthy subjects and patients [17,18]. If the predicted PK profile and parameters significantly deviated from the observed data, the model would then be refined by parameter optimization by fitting against the observed clinical data. Initial scaling using enzyme kinetics values for both enzymes, determined in vitro, significantly under-predicted in vivo CLORAL. The enzyme kinetics parameters were, therefore, optimized using the GastroPlus build-in optimization moduleTM, in order to capture the PK profile of chlorzoxazone in healthy volunteer clinical studies. In addition, the Kp values for all tissues were optimized using GastroPlus, in order to fit the plasma profiles and to match the observed Vss. The verified PBPK model was then used to evaluate the effect of diabetes mellitus on the dynamics of chlorzoxazone.
Development and verification of PBPK model in diabetes mellitus: In the present study, we simulated the PK of tested compounds in subjects with diabetes mellitus using the PBPK models. The diabetes mellitus PBPK (DM-PBPK) model incorporates physiological parameters, physiochemical properties of the tested compounds, ADME data, and information obtained from clinical studies. The DM-PBPK model was developed by taking into consideration the physiological changes that occur in DM (e.g., changes in the absorption, changes in the protein binding and distribution, and CL changes contributed by CYP450 enzymes). Table 3 summarizes all of the changes that occur in diabetes mellitus which may have direct impact on the pharmacokinetics of the tested compounds. Changes in gut physiology, including delayed gastric emptying time and slow transient times in the gastric system and small intestine, were applied directly to the ACAT models, in order to reflect the changes in the absorption rate of the tested compounds. The hepatic uptake of the tested compounds was kept constant, due to the lack of reports investigating the effect of DM on the hepatic uptake of drugs. Various physiological changes in DM patients can alter the volume of distribution of drugs, mainly affected by changes in body weight and protein binding. Obesity, which is very common in diabetic patients, directly affects the Vd of lipophilic drugs. In the proposed DM-PBPK model, the DM subjects were considered to be obese in the construction of the models for the tested compounds. In addition, both compounds have high protein binding and, so, are affected by changes in the protein binding (decreased by 10%) during DM [2]. Drug elimination is significantly altered during pregnancy due to alterations in metabolic enzyme activities. The change in hepatic enzyme activity in DM is CYP-isoform specific. Previous studies have shown that CYP3A4 activity is down-regulated significantly in diabetic patients [2]. Meanwhile, the opposite occurs for CYP3A5 and CYP2E1. The predicted mean PK parameters (Cmax, AUC) for diabetic and non-diabetic subjects were obtained based on simulations.
Table 3. PK changes in diabetes
|
Diabetic |
Reference |
Absorption |
|
Gastric mucosal blood flow |
↓ 50% |
[27] |
Gastric emptying |
↓ 35–72% |
[28, 29] |
Gastric transit time |
↓ 70% |
[30] |
Small bowel transit time |
↓ 40–50% |
[31] |
Muscle blood flow |
↓ 20% |
[32] |
Distribution |
|
Protein binding |
↓ 10% |
[2] |
Metabolism |
|
CYP2E1 |
↑ 50% |
[2] |
CYP3A4 |
↓ 40–80% |
[2] |
CYP3A5 |
↑ 30% |
[2] |
CYP1A2 |
↓ 15–45% |
[2] |
UGT1A1 |
↓ 40% |
[2] |
UGT1A9 |
↓ 45% |
[2] |
UGT2B7 |
↓ 75% |
[2] |
Results
Glibenclamide: The PBPK model of glibenclamide in non-diabetic subjects was developed using reported values for individual CYP enzyme metabolism, free fraction in plasma, blood-to-plasma ratio, effective permeability, and physicochemical parameters (Table 1). The constructed PBPK model was first verified against reported disposition kinetics obtained from a single dosing study, and then further verified against the disposition kinetics of another set of single dosing studies. The simulated plasma concentration–time profiles captured the observed PK data (Figure 2a, 2b). Model-predicted AUC and Cmax met the verification criterion, with predicted/observed ratio in the range of 0.8–1.2 (Table 4).
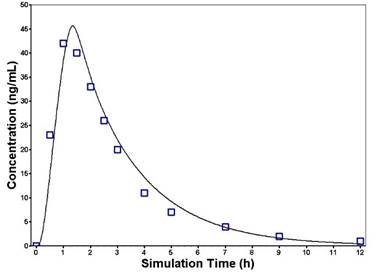
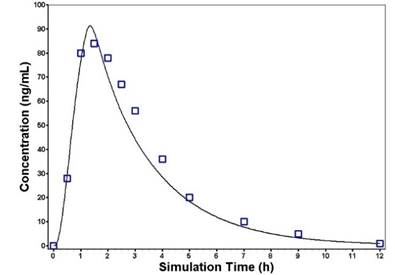
Figure 2. Predicted and observed plasma concentration–time profiles of glibenclamide following oral administration of a single dose of 0.875 mg (a) or 1.75 mg (b). The solid line represents the predicted mean glibenclamide profile. Mean observed (symbols) data are overlaid [12,13]
Table 4. Glibenclamide PK parameters after oral dosing in healthy and diabetic subjects
Dose |
Subjects |
Cmax (ng/mL) |
Fold error |
AUC (ng.h/mL) |
Fold error |
Observed |
Predicted |
Observed |
Predicted |
0.875 mg PO |
Healthy |
42 |
45.7 |
1.08 |
137.3 |
136.4 |
0.99 |
1.75 mg PO |
Healthy |
84 |
91.2 |
1.08 |
312.3 |
272.9 |
0.87 |
2.5 mg PO |
Diabetic |
69 |
70 |
1.07 |
44 |
47 |
1.04 |
10 mg PO |
Diabetic |
240 |
281.9 |
1.17 |
1724.9 |
1879.2 |
1.08 |
After establishing and verifying the PBPK model in non-diabetic subjects, the DM-PBPK model was constructed by incorporating all physiological changes during diabetes. The simulated plasma concentration–time profiles based on the DM-PBPK model captured the observed PK data in diabetic subjects (Figure 3). Again, the predicted AUC and Cmax were comparable with the observed values, with predicted-to-observed ratio in the range of 0.8–1.2 (Table 4).
Chlorzoxazone: The PBPK model of chlorzoxazone in non-diabetic subjects was developed using reported values for individual CYP enzyme metabolism, free fraction in plasma, blood-to-plasma ratio, effective permeability, and physicochemical parameters (Table 2). The constructed PBPK model was first verified against reported disposition kinetics obtained from a single dosing study. The simulated plasma concentration–time profiles were similar to the observed PK data (Figure 4). Model-predicted AUC and Cmax met the verification criterion, with predicted/observed ratio in the range of 0.8–1.2 (Table 4). After establishing and verifying the PBPK model in non-diabetic subjects, the DM-PBPK model was built by incorporating all physiological changes during diabetes. Again, predicted AUC and Cmax were comparable with the observed values, with predicted-to-observed ratio in the range of 0.8–1.2 (Table 5).
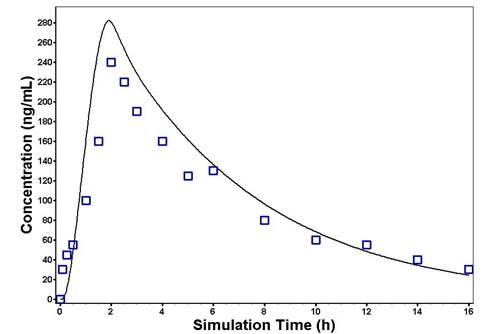
Figure 3. Predicted and observed plasma concentration–time profiles of glibenclamide following oral administration of a single dose of 10 mg in diabetic subjects. The solid line represents the predicted mean glibenclamide profile. Mean observed (symbols) data are overlaid [12,13]
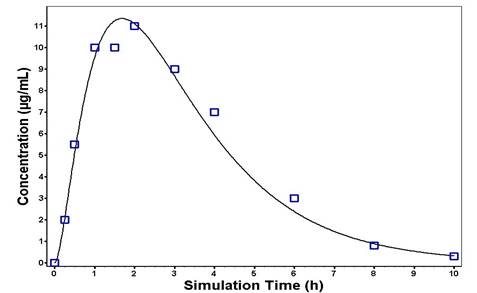
Figure 4. Predicted and observed plasma concentration–time profiles of chlorzoxazone following oral administration of a single dose of 50 mg in healthy subjects. The solid line represents the predicted mean chlorzoxazone profile. Mean observed (symbols) data are overlaid [17,18]
Table 5. Chlorzoxazone PK parameters after single oral dosing in healthy and diabetic subjects
Dose |
Subjects |
Cmax (ng/mL) |
Fold error |
AUC (ng.h/mL) |
Fold error |
Observed |
Predicted |
Observed |
Predicted |
500 mg PO |
Healthy |
11 |
11.3 |
1.01 |
48.8 |
45.7 |
0.93 |
500 mg PO |
Diabetic |
3.2 |
3.3 |
1.01 |
13.9 |
13.8 |
0.99 |
Discussion
The goal of this work was to develop a PBPK model to predict the pharmacokinetic behaviors in diabetic patients by incorporating knowledge of changes in many parameters associated to diabetes disease, such as stomach and intestinal transit time, renal function, and hepatic enzyme activities.
The PBPK model is a mathematical model that integrates drug and physiology data to simulate the pharmacokinetic profile of a drug in plasma and tissues. Physiological parameters are usually altered under disease conditions such as diabetes. This include changes in body weight, organ weights, blood flow rates, gut transit time, and enzyme activities (Table 3). The best method for testing how these factors affect the PK of medications is through PBPK modeling.
Since it is unlikely that the PBPK model parameters used for healthy populations will apply to diabetic individuals, the variations to these physiological factors brought on by diabetes should be incorporated into the PBPK model. The accuracy of the PBPK model's predictions of the pharmacokinetic characteristics of two selected drugs (glibenclamide and chlorzoxazone) in the healthy population. Then, the DM-PBPK model was developed, taking into consideration the physiological changes that occur in DM (e.g., changes in absorption, changes in protein binding and distribution, and CL changes contributed by CYP450 enzymes).
The PBPK model for glibenclamide and chlorzoxazone was developed using physicochemical, biopharmaceutical, and PK parameters obtained from the literature or estimated using the ADMET Predictor. The initial model was verified by comparing the simulated PK profiles with the observed data from several single and multiple dosing studies in healthy subjects and patients. The predicted pharmacokinetic parameters after a single oral dose or multiple oral doses were in agreement with clinical reports, with fold-errors within 0.8–1.2 (Tables 4 and 5). Then, the verified PBPK model was used to evaluate the effects of diabetes mellitus on the dynamics of glibenclamide and chlorzoxazone. We used this model to simulate the PK of the tested compounds in subjects with diabetes mellitus.
Eventually, the corresponding pharmacokinetic parameters of the two drugs in diabetic patients were further predicted using model parameters for corresponding diabetes patients. As a result, all clinical data were successfully predicted, with predicted mean population PK parameters (AUC and C max) falling within 80–120% of the observed values (Table 4 and 5). These results demonstrate that the PBPK model may accurately predict pharmacokinetic behavior in diabetes patients. The developed PBPK model, incorporating the alterations in various physiological and biochemical parameters induced by diabetes, was successfully used to predict the pharmacokinetic profiles of drugs in diabetic patients, according to information from the literature or a healthy population.
It remains difficult to anticipate accurately whether diabetes will affect a drug's pharmacokinetics. The hepatic clearance of some medications can be influenced by hepatic CYP activity and hepatic blood flow, and medications that are mostly eliminated through the kidneys can also be affected by renal function and renal clearance. Interest in how changes in pharmacokinetics and drug formulation in diabetes affect gut transit and intestinal absorption has recently grown. The fact that the present model takes into account how the changes in gut wall metabolism brought on by diabetes affect medication pharmacokinetics provides a significant advantage. Moreover, data on gut transit time revealed that sustained-release medicines and medications with poor intestinal absorption are significantly influenced by changes in gut transit time. However, more investigations of the physiological and functional changes that may occur in diabetic patients are required. In addition, more studies also need to be conducted to test the effects of these changes on the pharmacokinetic properties of various medications.
Conducting clinical investigations is never without ethical limitations. Chemically induced animal models of diabetes have been developed to get around these limitations. In animal models of diabetes, the impact of the disease on these features have been well-described. However, there is relatively little information available for humans, and it is unclear how the disease affects these properties. Nevertheless, it has been demonstrated that diabetes alters the pharmacokinetics and pharmacodynamics of medicines. At present, PBPK models and other modeling approaches could play a critical role in understanding such effects. The DM-PBPK model proposed in this work provides a feasible alternative to empirical dosage selection when no clinical studies providing guidance on dose recommendations in diabetic patients are available.
Conclusion
In vitro data and information from a healthy population were successfully used to predict the pharmacokinetic profiles of medications in diabetic patients using the developed DM-PBPK model, including changes in numerous anatomical, physiological, and biochemical characteristics caused by diabetes. When no clinical trials are available to guide dose recommendations in diabetic patients, the DM-PBPK model offers a workable substitute for empirical dosage selection.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project (number: RSP-2021/2), King Saud University, Riyadh, Saudi Arabia.
Conflicts of interest
The authors declare no competing interests for this work.
References
- Alotaibi A (2017) Incidence and prevalence rates of diabetes mellitus in Saudi Arabia: An overview. J Epidemiol Glob Health 7: 211-218. [Crossref]
- Dostalek M, F Akhlaghi, M Puzanovova (2015) Effect of diabetes mellitus on pharmacokinetic and pharmacodynamic properties of drugs. Clin Pharmacokinet 51: 481-499. [Crossref]
- Hempel A (1997) High glucose concentrations increase endothelial cell permeability via activation of protein kinase C alpha. Circ Res 81: 363-371.
- Ruiz-Cabello F, S Erill (1984) Abnormal serum protein binding of acidic drugs in diabetes mellitus. Clin Pharmacol Ther 36: 691-695.
- Baraka-Vidot J (2012) Impaired drug-binding capacities of in vitro and in vivo glycated albumin. Biochimie 94: 1960-1967. [Crossref]
- Dostalek M (2011) Significantly reduced cytochrome P450 3A4 expression and activity in liver from humans with diabetes mellitus. Br J Pharmacol 163: 937-947. [Crossref]
- Huang SM (2009) When to conduct a renal impairment study during drug development: US Food and Drug Administration perspective. Clin Pharmacol Ther 86: 475-479.
- Sasso FC (2006) Cardiovascular risk factors and disease management in type 2 diabetic patients with diabetic nephropathy. Diabetes Care 29: 498-503.
- Hanley MJ, Abernethy DR, Greenblatt DJ (2010) Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet 49: 71-87.
- Rodgers T and Rowland M (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95: 1238-1257.
- Zhou L (2010) Contributions of human cytochrome P450 enzymes to glyburide metabolism. Biopharm Drug Dispos 31: 228-242. [Crossref]
- Jönsson A (2000) Effects and pharmacokinetics of oral glibenclamide and glipizide in Caucasian and Chinese patients with type-2 diabetes. Eur J Clin Pharmacol 56: 711-714. [Crossref]
- Serra D (2008) Evaluation of pharmacokinetic and pharmacodynamic interaction between the dipeptidyl peptidase IV inhibitor vildagliptin, glyburide and pioglitazone in patients with Type 2 diabetes. Int J Clin Pharmacol Ther 46: 349-364.
- Rodgers T, Leahy D, Rowland M (2005) Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94: 1259-1276.
- Court, M.H (1997) Biotransformation of chlorzoxazone by hepatic microsomes from humans and ten other mammalian species. Biopharm Drug Dispos 18: 213-216. [Crossref]
- Kramer I (2003) Comparison of chlorzoxazone one-sample methods to estimate CYP2E1 activity in humans. Eur J Clin Pharmacol 59: 775-778.
- Wang Z (2003) Diabetes mellitus increases the in vivo activity of cytochrome P450 2E1 in humans. Br J Clin Pharmacol 55:77-85. [Crossref]
- Ono S (1995) Chlorzoxazone is metabolized by human CYP1A2 as well as by human CYP2E1. Pharmacogenetics 5:143-150.
- Wei H (2008) Physicochemical characterization of five glyburide powders: a BCS based approach to predict oral absorption. Eur J Pharm Biopharm 69: 1046-1056.
- Lobenberg R (2000) Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of glibenclamide. Pharm Res 17: 439-444. [Crossref]
- Hardman JG, Limbird LE (2001) Goodman & Gilman’s the Pharmacological Basis of Therapeutics. 2001.
- Olsen KM, Kearns GL, Kemp SF (1995) Glyburide protein binding and the effect of albumin glycation in children, young adults, and older adults with diabetes. J Clin Pharmacol 35: 739-745.
- Pearson JG (1985) Pharmacokinetics of glyburide. Am J Med 79: 67-71.
- Balant L (1981) Clinical pharmacokinetics of sulphonylurea hypoglycaemic drugs. Clin Pharmacokinet 6: 215-241. [Crossref]
- Morrison PJ (1982) Effect of pirprofen on glibenclamide kinetics and response. Br J Clin Pharmacol 14: 123-126.
- Cheng CL (2004) Biowaiver extension potential to BCS Class III high solubility-low permeability drugs: bridging evidence for metformin immediate-release tablet. Eur J Pharm Sci 22: 297-304.
- Zhu L 1(1993) Gastric mucosal blood flow and blood viscosity in patients with diabetes. Zhonghua Yi Xue Za Zhi 73: 476-478.
- Horowitz M, Fraser R (1994) Disordered gastric motor function in diabetes mellitus. Diabetologia 37: 543-551. [Crossref]
- Horowitz M (1996) Gastric emptying in diabetes: an overview. Diabet Med 13: S16-S22.
- Triantafyllou K (2007) Video-capsule endoscopy gastric and small bowel transit time and completeness of the examination in patients with diabetes mellitus. Dig Liver Dis 39: 575-580.
- Jung HK (2003) Colonic transit time in diabetic patients--comparison with healthy subjects and the effect of autonomic neuropathy. Yonsei Med J 44: 265-272. [Crossref]
- Leinonen H, Matikainen E, and Juntunen J (1982) Permeability and morphology of skeletal muscle capillaries in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 22: 158-162. [Crossref]