Abstract
Both diabetes types, types 1 and 2, are associated with cognitive impairments. Each period of life is concerned, and this is an increasing public health problem. Animal models have been developed to investigate the biological actors involved in such impairments. Many levels of the brain function (structure, volume, neurogenesis, neurotransmission, behavior) are involved. In this review, we detailed the part potentially played by the Hypothalamic-Pituitary Adrenal axis in these dysfunctions. Notably, regulating glucocorticoid levels, their receptors and their bioavailability appear to be relevant for future research studies, and treatment development.
Key words
type 1 diabetes, type 2 diabetes, HPA axis, glucocorticoids, memory, hippocampus
Introduction
Diabetes is characterized by the inability of the body to produce or respond to insulin, with the consequence that the body cannot control the level of sugar in the blood, namely glycemia. The latest edition of the International Diabetes Federation (IDF) Diabetes Atlas shows that more than 460 million adults are currently living with diabetes, with increasing prevalence, which represents a strong socio-economic burden. There are two main types of diabetes. Type 1 Diabetes (T1D) is commonly diagnosed during childhood and adolescence and is characterized by the total lack of pancreatic cells producing insulin. T1D represents 5% of all diabetes types. Type 2 diabetes (T2D) is more common, often develops later in life, and is usually associated with metabolic syndrome. In T2D, high plasma glucose level is due to a default of insulin secretion from the pancreas, or a default in insulin action (insulin resistance). Diabetes shows comorbidities such as cancer, infections, cardiovascular conditions or mental / cognitive disorders. Moreover, diabetes induces a background vulnerability that can facilitate the negative effects of these comorbidities [1]. This suggests an additive and/or synergistic relationship between risk factors of other diseases, consistent with the “diathesis” hypothesis of diabetes [2].
In the organism, the brain uses almost 20% of total plasma glucose. Glucose uptake in the brain is mainly insulin-dependent, and insulin receptors are widely distributed in the brain [3]. Brain function critically depends on glucose supply and insulin in the brain regulates several functions, such as whole-body energy metabolism in the hypothalamus, or memory formation in the hippocampus [4]. At childhood and adolescence, glucose requirement is increased for brain growth and development. Conversely, prolonged hyperglycemia induces neuronal damages in both humans and other mammals. In rodents, elevated glycemia imped myelin formation and neurotransmission [3], which can be critical according to the state of development. The term “diabetic encephalopathy” was introduced in 1950 as the expression of the central complications of diabetes [5,6].
Processes linking diabetes to cognitive dysfunctions have been recently investigated and the Hypothalamic-Pituitary Adrenal (HPA) axis appears as a key player in these processes. Here, we aimed to review cognitive impairments induced by diabetes, and the involvement of a high HPA axis in the cognitive dysfunctions induced by T1D and T2D.
Memory impairment in Diabetes
T1D:
Biessels et al. [7] suggest a ‘‘critical periods hypothesis” whereby neurocognitive deficits in T1D occur predominantly at two crucial periods of life, when the brain is developing in early childhood and when the brain undergoes neurodegenerative processes with ageing. Of note, the two positions are not mutually exclusive.
T1D occurs most often during childhood in humans when brain development is important. This implies that cognitive impairments following T1D can arise early in life [8]. Recently, Liu et al. showed that childhood-onset T1D was associated with an increased risk of neurodevelopmental disorders including attention-deficit/hyperactivity disorder, autism spectrum disorders and intellectual disability in a Swedish cohort [9]. Northam et al. [10] showed in children with T1D, deficits in general intelligence, speed of processing, learning capacity, attention, processing speed, long-term memory, and executive skills [11]. Cognitive dysfunctions are also observed in adolescents with T1D, impaired abilities of planning, adapting and reacting to environment, especially in concept formation, cognitive flexibility, anticipation, problem-solving capacity and word reading speed [12] [13,14]. In these studies, T1D-induced cognitive alterations are independent of the quality of metabolic control and disease duration. In parallel, recent studies found associations between poor metabolic control and lower academic achievement, school performance, problem-solving capacity and cognitive flexibility [15-17], correlated with abnormal MRI data [18]. Aye et al. showed that a history of diabetic ketoacidosis was associated with changes in longitudinal cognitive and brain development [19]. Using a cross-sectional study design, cognitive and academic tests showed that young T1D subjects had lower verbal intelligence level compared to controls [17]. Increased exposure to hyperglycemia was associated to their lower spelling performance. Schoenle et al. [20] showed that impaired intellectual development in children with T1D was associated with glycated hemoglobin (HbA1c), age of diagnosis and sex.
Extreme hyperglycemic episodes, conducting to ketoacidosis (high levels of blood acids called ketones), affect strongly the brain [21], but chronic hyperglycemia has also been shown to be deleterious [22-25]. Acute hyperglycemia negatively affected spatial working memory in adolescents [26], and chronic hyperglycemia is associated with smaller volume of grey matter in the right cuneus and precuneus, smaller white matter volume in the right posterior parietal region, and larger grey matter volume in the right prefrontal region [23,27]. In young children (4-10 years of age), alteration of gray matter related to hyperglycemia [28] have been showed in regions with rapid development, such as bilateral occipital and cerebellar regions, left inferior prefrontal cortex, insula and temporal pole region. For Cameron et al. [8] the cerebral consequences of a history of diabetic ketoacidosis and chronic hyperglycaemia appear to have been underestimated compared to hypoglycemia in children.
Adult patients suffering from T1D with poor metabolic control (Hba1c > 8.8 %) show a psychomotor decline compared to those with better control (Hba1c < 7.4 %) [6]. In a prospective study Ryan et al. [5] found that adults with T1D showed significant declines on measures of psychomotor efficiency compared to non-diabetic controls. Nevertheless, no difference was seen in the domains of learning, memory, or problem-solving tasks. At the same time, cross-sectional studies have shown that T1D subjects have performance deficits in multiple cognitive domains including information processing speed, psychomotor efficiency, memory, attention, academic achievement, visuospatial abilities and executive function [6,29]. Current and information processing abilities are poorer in young adults with early onset of diabetes (<7 years old) than those with later onset (7-17 years old) [30]. Nunley et al. [31,32] found in adult diabetic patients an incidence rate of cognitive impairments of 28% in comparison with 5% in controls. Even if cognitive deficits appear mild to moderate, they might hamper the day-to-day life, by limiting action of patients in more demanding situations [33]. Cognitive dysfunction appears inversely related to HbA1c, i.e. appropriate glucose control [34].
Diabetic brain features several symptoms best described as “accelerated brain ageing” as suggested by Biessels et al. [11]. Reduced white matter volume was associated with decreased cognitive performance by Wessels et al. [29], and microvascular complications such as retinopathy were also predictive of cognitive dysfunction. MRI measurements were not conclusive concerning a potential cerebral atrophy [33], nevertheless early diabetes onset has been associated with a higher ventricular volume [30,35].
The prolonged life span in T1D induces a risk for aging-related disease as dementia [32]. A retrospective study estimated that the risk ratio for dementia in T1D patients was 1.65 times compared to non-T1D subjects (32]. Neurodegenerative state interferes with daily life activities and increase the dependency of T1D patients to social assistance [32,36].
For McCrimmon, hypoglycemia and its rebound hyperglycaemia interact synergistically to improve oxidative stress and inflammation, damaging vulnerable brain regions and accelerating cognitive decline [37]. Poor management of hypoglycemia episodes is associated with smaller gray matter volume in the frontal lobe of children [27]. Kodl et al. [38] showed a correlation between reduced fractional anisotropy (representing neuronal connectivity), the duration of diabetes, and cognitive impairment in young adults. However, recurrent hypoglycemia, even severe, was not correlated with fractional anisotropy. Severe hypoglycemia was associated with smaller grey matter volume in the left superior temporal region [25]. T1D children experiencing severe hypoglycemia episodes before the age of five show deficits like delayed recall and spatial orientation skills [25] [39]. Kaufman et al. [22] showed that stable glycemia improves cognitive abilities in young children. The hippocampus, a brain structure highly involved in spatial learning, is especially vulnerable to damage induced by hypoglycemia [40,42]. This provides a potential mechanism for learning and memory dysfunction occurring with T1D [43]. In T1D adults, when severe hypoglycemia occurs (seizures, even coma), studies have shown impaired attention [44], verbal skills [45], short-term visual-spatial and verbal memory [22,46], vigilance [47], full-scale or verbal intelligence scores [12].
In rodents, a model of T1D has been developed by treatment with streptozotocin (STZ). STZ is a glucosamine-nitrosourea which is taken up specifically by Glut-2 glucose transporter, highly expressed in pancreatic beta-cells [48] and absent in the blood brain barrier [49]. Injection of STZ induces a significant and rapid destruction of pancreatic beta cells. STZ diabetic rodents are unable to produce insulin in sufficient quantities and suffer from exceedingly high glycemia (up to 600 mg/dl). STZ-diabetic rodents display polyuria, polydipsia and a strong delay in growth. In an ethical point of view, we do not advise researchers to keep STZ-T1D animals for a long time without insulin replacement [15,51]. For short diabetes duration (2-3 weeks), the measurement of plasma fructosamine concentration appears more relevant than HbA1c to characterize chronic hyperglycemia [51]. In STZ-T1D rat model, chronic hyperglycemia reduces neurons number and impedes myelination [52], and decreases spinogenesis and dendritic arborization inside the limbic system [53]. Both spatial learning and hippocampal long-term potentiation (LTP, reinforcement of synaptic contacts contributing to storage of information) are impaired, and enhancement of long-term depression (LTD, reduction of synaptic contacts causing the opposite effect of LTP) was measured in severely hyperglycemic rats. Both water-maze learning and hippocampal LTP are impaired in STZ diabetic rats [54]. Repetitive hypoglycemia in young rat also impairs hippocampal LTP [11]. Early treatment substitution with insulin reversed STZ effects on Morris water-maze and hippocampal LTP, but not late treatment (10 weeks) [54]. In rat, Malone et al. show that chronic hyperglycemia could be more damaging to the developing brain than intermittent hypoglycemia [52], whereas this observation is still debated in human. Nevertheless, hypoglycemia was shown by Suh et al. [40] to induce transient neurogenesis (2 weeks) and subsequent progenitor cell loss (4 weeks) in the rat hippocampus, via a sustained activation of glutamate receptors in dentate gyrus. Studies in diabetic rats have suggested that ketoacidosis-induced chronic neuroinflammation could contribute to the cognitive decline of T1D individuals [55].
Recently, intracerebroventricular STZ injection (3 mg/kg) was shown to decrease cerebral glucose uptake (in particular in hypothalamus and circumventricular organs), and to produce multiple effects that mimic molecular, pathological and behavioral features of Alzheimer’s disease (AD) [56]. This inability to metabolize glucose, and by extension AD linked to diabetes, have been called Type 3 Diabetes (T3D) [57]. STZ administration in rat lateral ventricles decreases brain glucose use in the frontal and parietal cortex, and by reducing ATP and phosphocreatine availability, diminishes energy charge potential in the cerebral cortex
[21].
There is a strong association between T1D and cognitive impairments. It becomes more and more relevant to define the risk factors involved in this complication, and to search therapeutic targets on which we could act.
T2D:
T2D is one of the most common chronic metabolic diseases. As a result of high calorie diets and sedentary lifestyles, diabetes is rapidly becoming more prevalent in Western societies. While global prevalence of diabetes in urban areas is 10.8%, in rural areas it is lower, at 7.2%.
However, this gap is closing, with rural prevalence on the rise (IDF Diabetes atlas, 9th edition 2019). In addition to its well–known adverse effects on the cardiovascular and peripheral nervous systems, T2D also appears to negatively impact the brain, increasing the risk of depression and dementia [59]. Prospective studies showed that T2D people perform less in verbal memory, memory, information-processing speed, attention and executive function [60-62], in association with an hippocampal atrophy [27]. Mental flexibility and global cognition were not affected in all studies [63]. Cognitive decrements were associated with early onset of diabetes and poor glycemic control (ACCORD Memory in Diabetes study, MIND) [64]. Both genetic and environmental factors such as a lack of exercise, obesity, smoking, stress, and aging affect the development of insulin resistance which is involved in neurodegeneration [65,66]. MRI analyses identified structural atrophy (white, total gray matters, and volume of hippocampus [67], cortex or amygdala [68,69], associated with neuropsychological deficits and vulnerability to dementia [70,71].
Experimental animal models of T2D show impairments in hippocampal-based memory performance [72], deficits in hippocampal neuroplasticity including decreases in dendritic spine density and neurogenesis [59] and decreases in synaptic transmission (LTP) [73], whereas bolstering insulin signaling mitigates β-amyloid-induced synapse loss in mature cultures of hippocampal neurons [72]. In a model of hippocampal-specific insulin resistance, rats showed deficits in LTP and spatial memory, especially long-term memory [74]. Central insulin administration improves spatial memory in a dose-dependent fashion in male rats [75], whereas intrahippocampal insulin microinjections enhances memory consolidation and retrieval [76]. Acute delivery of insulin into the rat hippocampus also promotes spatial memory in the alternation test [77], and transiently enhances hippocampal-dependent memory in the inhibitory avoidance test [78].
Aging is usually associated to insulin resistance, T2D and cognitive decline. Moreover, T2D was found associated with 50% increased risk of dementia [79,80], AD and vascular disorders [81]. Diabetes is now considered to be the second leading risk factor for AD, following aging itself [82]. Interestingly, Smolina et al. [36] measured an increased ratio for developing dementia in both diabetes (T1D and T2D) in a large cohort of patients in England.
T2D is often associated to weight excess. Proinflammatory cytokines produced by adipocytes can cross the blood-brain barrier and induce neuroinflammation, which favors subsequent neurodegeneration. Dietary manipulations appeared to ameliorate T2D alterations in periphery that were shown to be epidemiologically linked to a decreased incidence of AD and to retard pathogenesis in animal models of T2D [82]. Increased inflammation induces accelerated Aβ deposition [83] and/or decreased clearance and facilitates the polymerization of Tau. A recent study suggests that Tau could be preferentially involved in synaptic and cognitive deficits in T1D than in T2D experimental models [84]. Insulin signaling disorders also promote neuro-inflammation (notably elevated levels of cytokines/chemokines and gliosis [85], apoptosis, oxidative stress, impairments of energy metabolism and synaptic disconnections [86], all of which lead to the development of cognitive impairment and AD, prompting some investigators to refer AD as T3D, characterized by an insulin-resistant brain state [87]. The concept of brain insulin resistance was advanced by Arnold et al. [88]. Interestingly, the brain regions with the highest densities of insulin receptors, such as the hippocampus and temporal lobe, are also the major targets of neurodegeneration in AD. Studies showed a strong genetic association between T2D and AD, with candidate genes involved in insulin resistance, and others leading to uncontrolled inflammatory stress on neuronal tissues, which can precipitate the formation of amyloid and Tau proteins, thus worsening AD development [89,83]. Recently, Kshirsagar et al. reviewed the role played by insulin resistance as the connecting link between AD and T2D [90).
To conclude, drugs that increase insulin sensitivity might have a positive effect on the cognitive consequences of diabetes. In this regard, members of the incretin family (notably Glucagon-like peptide-1 (GLP-1), exendin-4 or liraglutide) could be considered as prime candidates to delay cognitive decline, and even to improve the mild cognitive decline and AD symptoms [4,72]. Recently, brain derived neurotrophic factor (BDNF), dipeptidyl peptidase4 (DPP4), and Ca2+ brain dysregulation were identified as novel biomarkers and potential therapeutic targets for diabetes-related cognitive impairment [91,92].
An intranasal treatment with insulin could be a promising perspective to prevent cognitive deficiencies induced by diabetes regardless of patient age. Insulin via this route of administration travels in convective bulk flow along perivascular pathways following the olfactory and trigeminal nerves and importantly by passing the blood brain barrier. In this way, insulin can reach the hippocampus and the cortex in 15–30 min [93,94]. Importantly, intranasal insulin does not reach the general circulation [95], thereby avoiding peripheral hypoglycemia. Intranasal insulin administration improves working memory in both human and animal studies, and intrahippocampal delivery of insulin improves hippocampal-dependent spatial working memory. This positive effect was due to regional vasoreactivity, especially vasodilatation in the anterior brain regions, such as insular cortex that regulates attention-related task performance [4]. Moreover, by inhibiting apoptosis, insulin promotes neuronal survival [88]. Novak et al. [96] demonstrated that intranasal insulin increases resting-state connectivity between the hippocampus and the medial frontal cortex compared to placebo.
Involvement of HPA axis in memory impairments induced by diabetes
Glucocorticoids (cortisol in humans and most mammals, corticosterone in rats and mice) are produced by the adrenal cortex and regulated by ACTH under the control of the HPA axis [97]. The major determinants of corticosteroid action are the level of free cortisol in the plasma and the densities of its receptors in target tissues. As little as 5% of cortisol circulates free in the plasma, with the majority bound with high affinity to corticosteroid binding globulin, which acts as a reservoir and a transporter of steroid to target cells, as well as lower affinity proteins such as albumin. The production of glucocorticoids is contingent upon the pronounced circadian rhythm: low during quiescence/sleep (2-10 ng/ml in male rat; 5-20 ng/ml in human), and high during the active phase (100-250 ng/ml in male rat; 50-150 ng/ml in human). Episodic stressful events stimulate HPA axis which can strongly increase glucocorticoids in blood (up to 200-300 ng/ml in male rat, and up to 500 ng/ml in human), and considerable variations in free plasma cortisol concentrations occur. Tissue glucocorticoid action is also determined by an intracellular enzyme that metabolizes glucocorticoids, the 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1, [98]). Cortisol is believed to diffuse across cell membranes and then to bind to cytoplasmic glucocorticoid receptors (GR), which then translocate to the nucleus. GR are ligand-gated transcription factors that regulate a plethora of genes directly or through interactions with other transcription factors. In some tissues, especially those involved in memory, glucocorticoids also bind mineralocorticoid receptors (MR) with a higher affinity than GR.
Due to their pivotal role in endocrine systems, glucocorticoids have been associated to vulnerability to many diseases, and to pathology complications. Taking into account the involvement of the HPA axis in neurobiology and memory, impairments in HPA function may exacerbate the neurobiological complications of both T1D and T2D [99].
Glucocorticoid levels and cognitive functions
Both absence and excessive levels of glucocorticoids appear deleterious for cognitive functions. Removal of glucocorticoids by adrenalectomy or synthesis inhibition by metyrapone impairs memory consolidation in rats [100,101], i.e basal levels of glucocorticoids are needed for neuronal maintenance. Conversely, elevated cortisol levels have been associated with poor cognitive ability in humans subjected to psychosocial stress [102], during normal aging [103] and in AD [104]. In rats, high glucocorticoids induces morphological changes in hippocampus (notably in CA3), and cognitive deficits [105]. An acute stressful experience decreases the number of adult-generated neurons in the dentate gyrus in various species, and hippocampal volume [106]. Moreover, chronic exposure to stress-induced elevations in corticosterone suppresses adult neurogenesis, reduces LTP, and impairs learning on hippocampus-dependent tasks in rats, such as behavior in the Y-maze [107-109]. Diamond et al. [110] reported an inverted U-shape relationship between the level of circulating corticosteroids, and LTP in the hippocampus of rats. The induction of LTP in the hippocampus is blocked by the administration of corticosterone, and there is a negative correlation between the magnitude of LTP in the hippocampal CA1 and the level of circulating corticosteroids [106]. According to Roozendal et al., glucocorticoid effects on memory also depends on catecholamines, notably in the basolateral amygdala [111]. Related to diabetes, it has been shown that memory deficits induced by chronic restraint stress are related to an impaired insulin signaling in mice, that is rescued by intranasal insulin treatment [112].
T1D
Humans with poorly controlled diabetes exhibit disturbances in their HPA axis function, resulting in increased basal activity, high nocturnal rise in plasma cortisol, impaired negative feedback [113], and greater responses to CRH stimulation [114]. Similarly, levels of adrenal glucocorticoids are elevated in rodents with experimental diabetes, which has been linked to memory impairments and underlying dendritic reorganization [115]. These alterations are reversed by a return to basal glucocorticoid concentrations [108], or prevented by inhibiting glucocorticoid synthesis [116]. Importantly, these alterations are also reversed with insulin treatment [117]. Because glucocorticoid excess has been causally linked to decreased hippocampal neurogenesis and cognitive deficits in STZ-induced diabetic adult rodents, it was important to evaluate glucocorticoid regulation in juvenile models of T1D, regarding the mean age of occurrence of T1D in human. Elevated plasma concentration of corticosterone was detected in untreated diabetic rats [118], as expected with such a chronic metabolic stress, but only at the nadir of secretion [51]. In response to a restraint stress, the peak of secretion of corticosterone was similar between experimental groups, contrary to the recovery that was delayed in untreated diabetic rats [51], suggesting an impaired feedback mechanism as described in humans. Although insulin treatment prevented the increased levels of corticosterone in basal conditions, there was no beneficial effect under stress conditions [50]. Within hippocampus, corticosterone concentration was also increased at the nadir in untreated diabetic rats, as well as 11β-HSD1 activity. The impaired negative feedback observed after stress may involve an altered neuronal environment that could be related to microstructural and neurogenesis data [51]. It has been suggested that chronic high glucocorticoid levels are responsible for the functional and morphological degeneration occurring in the hippocampus of T1D rats and mice [59]. Indeed, Stranahan et al. [119] reported that impaired LTP, deficits in cognitive performance and reduced neurogenesis were normalized in diabetic rodents after adrenalectomy and corticosterone replacement at physiological concentrations [120].
T2D
High glucocorticoid secretion is associated with T2D, and promotes gluconeogenesis in the liver, suppresses peripheral glucose uptake, enhances lipolysis, decreases insulin secretion in parallel with an increase in insulin resistance and inflammation [121]. Furthermore, the hippocampal insulin resistance elicited by corticosterone may contribute to the deleterious consequences of hypercortisolism/hyperglycemia observed in T2D [122], i.e. deficits in hippocampal neurogenesis, synaptic plasticity and learning. Such deficits are observed in db/db mouse, a model of T2D [59] in which obesity, hyperglycemia, and elevations in circulating corticosterone levels arise from a mutation that inactivates the leptin receptor [120]. Stranahan et al. [119] studied adrenalectomized db/db mice that were given different doses of corticosterone via drinking water (25 or 250 μg/ml in 0.9% saline). Adrenalectomized db/db mice that had received 25 μg/ml corticosterone replacement learned the location of the hidden platform in the Morris water maze more rapidly than sham–operated db/db mice and adrenalectomized db/db mice receiving 250 μg/ml corticosterone replacement.
In T2D patients, Bruehl et al. [99] measured an increased responsiveness to CRH, coupled with diminished suppression after dexamethasone-responsive test, indicating an abnormality in HPA feedback sensitivity (suspected to be a predictive factor of T2D, [123]), and related to decreased cognitive performance [124], but the brain pathways that mediate these links have not been understood yet. Reynolds et al. [125] showed that morning cortisol levels in elderly people with T2D are high, with deleterious effects on cognitive function. It has moreover been demonstrated that intranasal insulin may normalize stress axis activity in humans by reducing cortisol levels [4]. This inhibitory effect may also contribute to its positive impact on cognitive function. Glycogen synthase kinase 3 ß promotes Tau phosphorylation which impairs memory in T2D and is involved in AD, and has been shown to be activated by glucocorticoids [126].
Bioavailability of glucocorticoids: the enzyme 11ß-HSD1
The intracellular enzyme 11ß-HSD1 catalyzes intracellular regeneration of active glucocorticoids from inert 11-keto forms in liver, adipose tissue and brain [127]. It is widely expressed in hippocampus, cerebellum, and neocortex, suggesting its potential involvement in processes such as memory and learning. Sandeep et al. [128] showed that oral administration of an 11ß-HSD1 inhibitor, carbenoxolone, improved verbal fluency in 10 healthy elderly men and improved verbal memory in 12 T2D patients. Quervain et al. showed that a rare haplotype in the 5′ regulatory region of the gene encoding 11ß-HSD1 was associated with an increased risk to develop AD [129]. Related to T1D, our group showed elevated level of 11β-HSD1 in the morning urine of T1D children, even in the presence of insulin treatment [130]. This result suggests an alteration of glucocorticoids metabolism associated to T1D.
The important role of hippocampal 11β-HSD1 has recently been demonstrated in a rodent model of aging [131,132]. Indeed, 11ß-HSD1 null-mice resist to cognitive decline with aging [127]. Furthermore, treatment of aged rodents with liquorice [133], carbenoxolone, or synthetic selective inhibitors of 11ß-HSD1 (for instance UE2316 [131]) improves memory by decreasing local corticosterone concentration in brain [127,134,135] and by preventing brain atrophy [136]. Given our data in T1D children [137], we hypothesized that 11β-HSD1 may also be overactivated in brain of T1D and be responsible for cognitive impairments as it is the case in old individuals. We demonstrated that 11β-HSD1 levels were increased in T1D juvenile rats hippocampus [50,51]. These studies point to a pivotal role for 11β-HSD1 in glucocorticoid excess induced by T1D and consequently of altered hippocampal function (performance on the Y-maze and recognition of a displaced object [51]). An interesting perspective would be to evaluate the potential beneficial effect of 11β-HSD1 inhibition in untreated and insulin-treated diabetic juvenile rats to decipher the role of 11β-HSD1 in hippocampal-dependent cognitive alterations induced by T1D.
Receptors
Brain effects of glucocorticoids are mediated via two types of receptors: the mineralocorticoid receptors (MR) and the glucocorticoid receptors (GR). MR are highly and specifically expressed in hippocampus, basolateral amygdala and prefrontal cortex, a network of brain structures involved in memory and learning processes, while GR are widely expressed throughout the whole brain. Cortisol has a tenfold higher binding affinity for the MR (Kd 0.5 nM) than for the GR (Kd 15-20 nM). Consequently, MR are activated first when cortisol levels increase, followed by GR activation when cortisol levels increase further [138]. In accordance, it has been postulated that cortisol levels follow an inverted U-shaped dose response curve, with very low cortisol levels (predominantly activating MR), as well as very high cortisol levels (activating MR and a large amount of GR) negatively affecting the mediating function of these receptors on information processing (MR/GR balance) [139]. MR activation leads to retrieval of previously learned tasks and behavioral responses to new situations, is neuroprotective, and stimulates hippocampal function [140]. The literature suggests that MR mediate the role of corticosterone in the appraisal of novel situations, behavioral reactivity, and affective responses [100,141], and enhances the performance in spatial hippocampal-dependent cognitive tasks in rodents [142,143] and cognitive performances in human [144]. GR activation is responsible for the consolidation of new information [145], or of a stressful event [146]. A mutation preventing GR from dimerization suggests that gene transcription needs to be activated for the consolidation of memory [147]. Basal corticosterone levels are needed for effective LTP, but higher levels impair it and enhances LTD via GR. High glucocorticoids and stressors also suppress neurogenesis in the dentate gyrus. This U-shaped result may reflect the relative occupancy of MR (at lower doses) and GR (at higher ones) [146] [148]. Adrenalectomy also impairs consolidation as does the knockout of GR. However, cognitive performances are improved during chronic blockade of GR with the glucocorticoid antagonist mifepristone (RU486) [149,150].
Only few studies investigated the specific involvement of corticosteroid receptors in altered memory associated to diabetes. For instance, Revsin et al. [150] showed that GR blockade with mifepristone (RU486) improves hippocampal alterations and cognitive impairment in STZ-induced T1D in mice. Interestingly, polymorphisms on the MR and GRcoding genes were recently involved in cognition under stress [151] and AD [152]. Modulating specific activations and inhibitions of MR-/GR- might be a promising therapeutic way to prevent cognitive impairments induced by diabetes.
Conclusion
Targeting biological actors involved in HPA axis dysfunctions could be a relevant approach for the treatment of cognitive consequences of diabetes. Each period of life can be concerned. Figure 1 summarizes the vicious circle that can conduct to cognitive impairments, when brain consequences of diabetes are worsened by glucocorticoid-mediated effects, independently or not from aging or stressful conditions, and from variability of individual resilience (genetic and environmental vulnerability, comorbidities).
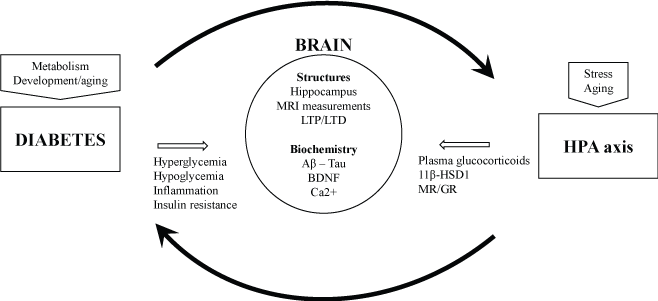
Figure 1. Potential biological targets for the therapeutic approaches of the cognitive impairments induced by diabetes. 11β-HSD1: 11β-Hydroxysteroid Deshydrogenase type 1; BDNF: brain derived neurotrophic factor ; HPA axis: Hypothalamic-Pituitary Adrenal axis ; LTP/LTD: long-term potentiation/depression ; MR/GR: mineralocorticoid/glucocorticoid receptors. MRI: Magnetic Resonance Imaging.
Acknowledgements
The authors thank Nicolas Mayer and Clementine Bosch-Bouju for critically reading the manuscript and helpful discussions.
References
- Gaudieri PA, Chen R, Greer TF, Holmes CS (2008) Cognitive function in children with type 1 diabetes: a meta-analysis. Diabetes Care 31: 1892–1897. [Crossref]
- Ryan CM (2006) Why is cognitive dysfunction associated with the development of diabetes early in life? The diathesis hypothesis. Pediatr Diabetes 7: 289–297. [Crossref]
- Fernandez AM, Torres-Alemán I (2012) The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci 13: 225–239. [Crossref]
- Benedict C, Grillo CA (2018) Insulin Resistance as a Therapeutic Target in the Treatment of Alzheimer’s Disease: A State-of-the-Art Review. Front Neurosci 12: 215. [Crossref]
- Ryan CM, Geckle MO, Orchard TJ (2003) Cognitive efficiency declines over time in adults with Type 1 diabetes: effects of micro- and macrovascular complications. Diabetologia 46: 940–948. [Crossref]
- Moheet A, Mangia S, Seaquist ER (2015) Impact of diabetes on cognitive function and brain structure. Ann N Y Acad Sci 1353: 60–71. [Crossref]
- Biessels GJ, Deary IJ, Ryan CM (2008) Cognition and diabetes: a lifespan perspective. Lancet Neurol 7: 184–190. [Crossref]
- Cameron FJ, Northam EA, Ryan CM (2019) The effect of type 1 diabetes on the developing brain. Lancet Child Adolesc Health 3: 427–436. [Crossref]
- Liu S, Kuja-Halkola R, Larsson H, Lichtenstein P, Ludvigsson JF, et al. (2021) Poor glycaemic control is associated with increased risk of neurodevelopmental disorders in childhood-onset type 1 diabetes: a population-based cohort study. Diabetologia 64: 767–777. [Crossref]
- Northam EA, Anderson PJ, Werther GA, Warne GL, Adler RG, et al. (1998) Neuropsychological complications of IDDM in children 2 years after disease onset. Diabetes Care 21: 379– 384.
- Biessels GJ, van der Heide LP, Kamal A, Bleys RLAW, Gispen WH (2002) Ageing and diabetes: implications for brain function. Eur J Pharmacol 441: 1–14. [Crossref]
- Northam EA, Anderson PJ, Jacobs R, Hughes M, Warne GL, et al. (2001) Neuropsychological profiles of children with type 1 diabetes 6 years after disease onset. Diabetes Care 24: 1541–1546. [Crossref]
- Brands AMA, Kessels RPC, de Haan EHF, Kappelle LJ, Biessels GJ (2004) Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels. Eur J Pharmacol 490: 159–168. [Crossref]
- Ohmann S, Popow C, Rami B, König M, Blaas S, Fliri C, Schober E (2010) Cognitive functions and glycemic control in children and adolescents with type 1 diabetes. Psychol Med 40: 95– 103. [Crossref]
- McCarthy AM, Lindgren S, Mengeling MA, Tsalikian E, Engvall J (2003) Factors associated with academic achievement in children with type 1 diabetes. Diabetes Care 26: 112–117. [Crossref]
- Kirchhoff BA, Jundt DK, Doty T, Hershey T (2017) A longitudinal investigation of cognitive function in children and adolescents with type 1 diabetes mellitus. Pediatr Diabetes 18: 443–449. [Crossref]
- Semenkovich K, Patel PP, Pollock AB, Beach KA, Nelson S, et al. (2016) Academic abilities and glycaemic control in children and young people with Type 1 diabetes mellitus. Diabet Med J Br Diabet Assoc 33: 668–673. [Crossref]
- Foland-Ross LC, Tong G, Mauras N, Cato A, Aye T, et al. (2020) Brain Function Differences in Children With Type 1 Diabetes: A Functional MRI Study of Working Memory. Diabetes 69: 1770–1778. [Crossref]
- Aye T, Mazaika PK, Mauras N, Marzelli MJ, Shen H, et al. Impact of Early Diabetic Ketoacidosis on the Developing Brain. Diabetes Care (2019) 42: 443–449. [Crossref]
- Schoenle EJ, Schoenle D, Molinari L, Largo RH (2002) Impaired intellectual development in children with Type I diabetes: association with HbA(1c), age at diagnosis and sex. Diabetologia 45: 108–114. [Crossref]
- Northam EA, Lin A, Finch S, Werther GA, Cameron FJ (2010) Psychosocial well-being and functional outcomes in youth with type 1 diabetes 12 years after disease onset. Diabetes Care 33: 1430–1437. [Crossref]
- Kaufman FR, Epport K, Engilman R, Halvorson M (1999) Neurocognitive functioning in children diagnosed with diabetes before age 10 years. J Diabetes Complications 13: 31–38. [Crossref]
- Perantie DC, Wu J, Koller JM, Lim A, Warren SL, et al. (2007) Regional brain volume differences associated with hyperglycemia and severe hypoglycemia in youth with type 1 diabetes. Diabetes Care 30: 2331–2337. [Crossref]
- Musen G, Jacobson AM, Ryan CM, Cleary PA, Waberski BH, et al. (2008) Impact of diabetes and its treatment on cognitive function among adolescents who participated in the Diabetes Control and Complications Trial. Diabetes Care 31: 1933–1938. [Crossref]
- Perantie Dana C, Lim Audrey, Wu Jenny, Weaver Patrick, Warren Stacie L, et al. (2008) Effects of prior hypoglycemia and hyperglycemia on cognition in children with type 1 diabetes mellitus. Pediatr Diabetes 9: 87–95. [Crossref]
- Šuput Omladič J, Slana Ozimič A, Vovk A, Šuput D, Repovš G, et al. (2020) Acute Hyperglycemia and Spatial Working Memory in Adolescents With Type 1 Diabetes. Diabetes Care 43: 1941–1944. [Crossref]
- Bednarik P, Moheet AA, Grohn H, Kumar AF, Eberly LE, et al. (2017) Type 1 Diabetes and Impaired Awareness of Hypoglycemia Are Associated with Reduced Brain Gray Matter Volumes. Front Neurosci 11: 529. [Crossref]
- Marzelli MJ, Mazaika PK, Barnea-Goraly N, Hershey T, Tsalikian E, et al. (2014) Neuroanatomical correlates of dysglycemia in young children with type 1 diabetes. Diabetes 63: 343–353. [Crossref]
- Wessels AM, Rombouts S a. RB, Remijnse PL, Boom Y, Scheltens P, et al. (2007) Cognitive performance in type 1 diabetes patients is associated with cerebral white matter volume. Diabetologia 50: 1763–1769. [Crossref]
- Ferguson SC, Blane A, Wardlaw J, Frier BM, Perros P, et al. (2005) Influence of an early-onset age of type 1 diabetes on cerebral structure and cognitive function. Diabetes Care 28: 1431–1437.
- Nunley KA, Rosano C, Ryan CM, Jennings JR, Aizenstein HJ, et al. (2015) Clinically Relevant Cognitive Impairment in Middle-Aged Adults With Childhood-Onset Type 1 Diabetes. Diabetes Care 38: 1768–1776. [Crossref]
- Li W, Huang E, Gao S (2017) Type 1 Diabetes Mellitus and Cognitive Impairments: A Systematic Review. J Alzheimers Dis 57: 29–36. [Crossref]
- Brands AMA, Kessels RPC, Hoogma RPLM, Henselmans JML, van der Beek Boter JW, et al. (2006) Cognitive performance, psychological well-being, and brain magnetic resonance imaging in older patients with type 1 diabetes. Diabetes 55: 1800–1806. [Crossref]
- Reaven GM, Thompson LW, Nahum D, Haskins E (1990) Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care 13: 16–21. [Crossref]
- Manolio TA, Kronmal RA, Burke GL, Poirier V, O’Leary DH, et al. (1994) Magnetic resonance abnormalities and cardiovascular disease in older adults. The Cardiovascular Health Study. Stroke 25: 318–327. [Crossref]
- Smolina K, Wotton CJ, Goldacre MJ (2015) Risk of dementia in patients hospitalised with type 1 and type 2 diabetes in England, 1998-2011: a retrospective national record linkage cohort study. Diabetologia 58: 942–950. [Crossref]
- McCrimmon RJ (2021) Consequences of recurrent hypoglycaemia on brain function in diabetes. Diabetologia 64: 971–977. [Crossref]
- Kodl CT, Franc DT, Rao JP, Anderson FS, Thomas W, et al. (2008) Diffusion tensor imaging identifies deficits in white matter microstructure in subjects with type 1 diabetes that correlate with reduced neurocognitive function. Diabetes 57: 3083–3089. [Crossref]
- Lin A, Northam EA, Rankins D, Werther GA, Cameron FJ (2010) Neuropsychological profiles of young people with type 1 diabetes 12 yr after disease onset. Pediatr Diabetes 11: 235–243. [Crossref]
- Suh SW, Fan Y, Hong SM, Liu Z, Matsumori Y, et al. (2005) Hypoglycemia induces transient neurogenesis and subsequent progenitor cell loss in the rat hippocampus. Diabetes 54: 500–509. [Crossref]
- Yamada KA, Rensing N, Izumi Y, De Erausquin GA, Gazit V, et al. (2004) Repetitive hypoglycemia in young rats impairs hippocampal long-term potentiation. Pediatr Res 55: 372–379. [Crossref]
- Hershey T, Perantie DC, Wu J, Weaver PM, Black KJ, et al. (2010) Hippocampal volumes in youth with type 1 diabetes. Diabetes 59: 236–241. [Crossref]
- Bjørgaas MR (2012) Cerebral effects of severe hypoglycemia in young people with type 1 diabetes. Pediatr Diabetes 13: 100–107. [Crossref]
- Rovet J, Alvarez M (1997) Attentional functioning in children and adolescents with IDDM. Diabetes Care 20: 803–810. [Crossref]
- Hershey T, Craft S, Bhargava N, White NH (1997) Memory and insulin dependent diabetes mellitus (IDDM): effects of childhood onset and severe hypoglycemia. J Int Neuropsychol Soc JINS 3: 509–520. [Crossref]
- Hershey T, Bhargava N, Sadler M, White NH, Craft S (1999) Conventional versus intensive diabetes therapy in children with type 1 diabetes: effects on memory and motor speed. Diabetes Care 22: 1318–1324. [Crossref]
- Howorka K, Pumprla J, Saletu B, Anderer P, Krieger M, Schabmann A (2000) Decrease of vigilance assessed by EEG-mapping in type I diabetic patients with history of recurrent severe hypoglycaemia. Psychoneuroendocrinology 25: 85–105. [Crossref]
- Schnedl WJ, Ferber S, Johnson JH, Newgard CB (1994) STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 43: 1326–1333. [Crossref]
- Kumagai Arno K (1999) Glucose transport in brain and retina: implications in the management and complications of diabetes. Diabetes Metab Res Rev 15: 261–273. [Crossref]
- Rougeon V, Moisan M-P, Barthe N, Beauvieux M-C, Helbling J-C, et al. (2017) Diabetes and Insulin Injection Modalities: Effects on Hepatic and Hippocampal Expression of 11β-Hydroxysteroid Dehydrogenase Type 1 in Juvenile Diabetic Male Rats. Front Endocrinol 8: 81. [Crossref]
- Marissal-Arvy N, Campas M-N, Semont A, Ducroix-Crepy C, Beauvieux M-C, et al. (2018) Insulin treatment partially prevents cognitive and hippocampal alterations as well as glucocorticoid dysregulation in early-onset insulin-deficient diabetic rats. Psychoneuroendocrinology 93: 72–81. [Crossref]
- Malone JI, Hanna SK, Saporta S (2006) Hyperglycemic brain injury in the rat. Brain Res 1076: 9–15. [Crossref]
- Flores-Gómez AA, de Jesús Gomez-Villalobos M, Flores G (2019) Consequences of diabetes mellitus on neuronal connectivity in limbic regions. Synap N Y N 73: e22082. [Crossref]
- Biessels G-J, Kamal A, Urban IJA, Spruijt BM, Erkelens DW, Gispen WH (1998) Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res 800: 125–135. [Crossref]
- Glaser N, Chu S, Hung B, Fernandez L, Wulff H (2020) Acute and chronic neuroinflammation is triggered by diabetic ketoacidosis in a rat model. BMJ Open Diabetes Res Care 8: [Crossref]
- Correia SC, Santos RX, Santos MS, Casadesus G, Lamanna JC, et al. (2013) Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Curr Alzheimer Res 10: 406–419.
- Kandimalla R, Thirumala V, Reddy PH (2017) Is Alzheimer’s disease a Type 3 Diabetes? A Critical Appraisal. Biochim Biophys Acta 1863: 1078–1089. [Crossref]
- Grieb P (2016) Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: in Search of a Relevant Mechanism. Mol Neurobiol 53: 1741–1752. [Crossref]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM (2008) Diabetes impairs hippocampal function via glucocorticoid–mediated effects on new and mature neurons. Nat Neurosci 11: 309–317. [Crossref]
- Hassing LB, Grant MD, Hofer SM, Pedersen NL, Nilsson SE, et al. (2004) Type 2 diabetes mellitus contributes to cognitive decline in old age: a longitudinal populationbased study. J Int Neuropsychol Soc JINS 10: 599–607. [Crossref]
- van den Berg E, Reijmer YD, de Bresser J, Kessels RPC, Kappelle LJ, et al. (2010) A 4 year followup study of cognitive functioning in patients with type 2 diabetes mellitus. Diabetologia 53: 58–65. [Crossref]
- Nici J, Hom J (2019) Neuropsychological function in type 2 diabetes mellitus. Appl Neuropsychol Adult 26: 513–521. [Crossref]
- Kanaya AM, Barrett-Connor E, Gildengorin G, Yaffe K (2004) Change in cognitive function by glucose tolerance status in older adults: a 4-year prospective study of the Rancho Bernardo study cohort. Arch Intern Med 164: 1327–1333. [Crossref]
- Launer LJ, Miller ME, Williamson JD, Lazar RM, Gerstein HC, et al. (2011) Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. Lancet Neurol 10: 969–977. [Crossref]
- Akhtar A, Sah SP (2020) Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem Int 135: 104707. [Crossref]
- Tyagi A, Pugazhenthi S (2021) Targeting Insulin Resistance to Treat Cognitive Dysfunction. Mol Neurobiol [Crossref]
- Moran C, Beare R, Wang W, Callisaya M, Srikanth V (2019) Type 2 diabetes mellitus, brain atrophy, and cognitive decline. Neurology 92: e823–e830. [Crossref]
- Li W, Huang E (2016) An Update on Type 2 Diabetes Mellitus as a Risk Factor for Dementia. J Alzheimers Dis JAD 53: 393–402. [Crossref]
- den Heijer T, Vermeer SE, van Dijk EJ, Prins ND, Koudstaal PJ, et al. (2003) Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 46: 1604–1610. [Crossref]
- Manschot SM, Brands AMA, van der Grond J, Kessels RPC, et al. (2006) Diabetic Encephalopathy Study Group. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 55: 1106–1113. [Crossref]
- Lee JH, Choi Y, Jun C, Hong YS (2014) Neurocognitive changes and their neural correlates in patients with type 2 diabetes mellitus. Endocrinol Metab Seoul Korea 29: 112–121. [Crossref]
- De Felice FG, Ferreira ST (2014) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63: 2262–2272. [Crossref]
- Kamal A, Ramakers GMJ, Gispen WH, Biessels GJ, Al Ansari A (2013) Hyperinsulinemia in rats causes impairment of spatial memory and learning with defects in hippocampal synaptic plasticity by involvement of postsynaptic mechanisms. Exp Brain Res 226: 45–51. [Crossref]
- Grillo CA, Piroli GG, Lawrence RC, Wrighten SA, Green AJ, et al. (2015) Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 64: 3927–3936. [Crossref]
- Haj-ali V, Mohaddes G, Babri SH (2009) Intracerebroventricular insulin improves spatial learning and memory in male Wistar rats. Behav Neurosci 123: 1309–1314. [Crossref]
- Moosavi M, Naghdi N, Choopani S (2007) Intra CA1 insulin microinjection improves memory consolidation and retrieval. Peptides 28: 1029–1034. [Crossref]
- McNay EC, Ong CT, McCrimmon RJ, Cresswell J, et al. (2010) Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem 93: 546–553. [Crossref]
- Stern SA, Chen DY, Alberini CM (2014) The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn Mem Cold Spring Harb N 21: 556–563. [Crossref]
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P (2006) Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5: 64–74. [Crossref]
- Zhang J, Chen C, Hua S, Liao H, Wang M, et al. (2017) An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract 124: 41–47. [Crossref]
- Kodl CT, Seaquist ER (2008) Cognitive dysfunction and diabetes mellitus. Endocr Rev 29: 494– 511.
- Griffith CM, Eid T, Rose GM, Patrylo PR (2018) Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology [Crossref]
- Sun Y, Ma C, Sun H, Wang H, Peng W, et al. (2020) Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J Diabetes Res 2020: [Crossref]
- Trujillo-Estrada L, Nguyen C, da Cunha C, Cai L, Forner S, et al. (2019) Tau underlies synaptic and cognitive deficits for type 1, but not type 2 diabetes mouse models. Aging Cell 18: e12919. [Crossref]
- Perry VH, Nicoll JAR, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6: 193. [Crossref]
- Sims-Robinson C, Kim B, Rosko A, Feldman EL (2010) How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 6: 551–559. [Crossref]
- Rorbach-Dolata A, Piwowar A (2019) Neurometabolic Evidence Supporting the Hypothesis of Increased Incidence of Type 3 Diabetes Mellitus in the 21st Century. BioMed Res Int 2019: 1435276. [Crossref]
- Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, et al. (2018) Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14: 168–181. [Crossref]
- Nisar O, Pervez H, Mandalia B, Waqas M, Sra HK (2020) Type 3 Diabetes Mellitus: A Link Between Alzheimer’s Disease and Type 2 Diabetes Mellitus. Cureus 12: [Crossref]
- Kshirsagar V, Thingore C, Juvekar A. (2021) Insulin resistance: a connecting link between Alzheimer’s disease and metabolic disorder. Metab Brain Dis 36: 67–83. [Crossref]
- Rozanska O, Uruska A, Zozulinska-Ziolkiewicz D (2020) Brain-Derived Neurotrophic Factor and Diabetes. Int J Mol Sci 21: [Crossref]
- Ma W-X, Tang J, Lei Z-W, Li C-Y, Zhao L-Q, et al. (2020) Potential Biochemical Mechanisms of Brain Injury in Diabetes Mellitus. Aging Dis 11: 978–987. [Crossref]
- Chapman CD, Frey WH, Craft S, Danielyan L, Hallschmid M, et al. (2013) Intranasal Treatment of Central Nervous System Dysfunction in Humans. Pharm Res 30: 2475–2484. [Crossref]
- Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG (2015) Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 35: 371–381. [Crossref]
- Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL (2002) Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci 5: 514–516. [Crossref]
- Novak V, Milberg W, Hao Y, Munshi M, Novak P, et al. (2014) Enhancement of Vasoreactivity and Cognition by Intranasal Insulin in Type 2 Diabetes. Diabetes Care 37: 751–759. [Crossref]
- Godoy LD, Rossignoli MT, Delfino-Pereira P, Garcia-Cairasco N, de Lima Umeoka EH (2018) A Comprehensive Overview on Stress Neurobiology: Basic Concepts and Clinical Implications. Front Behav Neurosci 12: [Crossref]
- Holmes MC, Seckl JR (2006) The role of 11β-hydroxysteroid dehydrogenases in the brain. Mol Cell Endocrinol 248: 9–14. [Crossref]
- Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E (2007) Hypothalamic-pituitary-adrenal axis dysregulation and memory impairments in type 2 diabetes. J Clin Endocrinol Metab 92: 2439–2445. [Crossref]
- Oitzl MS, de Kloet ER (1992) Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci 106: 62–71. [Crossref]
- Roozendaal B, Bohus B, McGaugh JL (1996) Dose-dependent suppression of adrenocortical activity with metyrapone: effects on emotion and memory. Psychoneuroendocrinology 21: 681– 693. [Crossref]
- Oei NYL, Everaerd WT a. M, Elzinga BM, van Well S, Bermond B (2006) Psychosocial stress impairs working memory at high loads: an association with cortisol levels and memory retrieval. Stress Amst Neth 9: 133–141. [Crossref]
- MacLullich AMJ, Deary IJ, Starr JM, Ferguson KJ, Wardlaw JM, et al. (2005) Plasma cortisol levels, brain volumes and cognition in healthy elderly men. Psychoneuroendocrinology 30: 505– 515. [Crossref]
- Elgh E, Lindqvist Astot A, Fagerlund M, Eriksson S, Olsson T, Näsman B (2006) Cognitive dysfunction, hippocampal atrophy and glucocorticoid feedback in Alzheimer’s disease. Biol Psychiatry 59: 155–161. [Crossref]
- Grillo CA, Piroli GG, Wood GE, Reznikov LR, McEwen BS, et al. (2005) Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience 136: 477–486.
- Lupien SJ, Lepage M (2001) Stress, memory, and the hippocampus: can’t live with it, can’t live without it. Behav Brain Res 127: 137–158. [Crossref]
- Gould E, Cameron HA, Daniels DC, Woolley CS, McEwen BS (1992) Adrenal hormones suppress cell division in the adult rat dentate gyrus. J Neurosci Off J Soc Neurosci 12: 3642–3650.
- Conrad CD, Lupien SJ, McEwen BS (1999) Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol Learn Mem 72: 39–46. [Crossref]
- Korz V, Frey JU (2003) Stress-related modulation of hippocampal long-term potentiation in rats: Involvement of adrenal steroid receptors. J Neurosci Off J Soc Neurosci 23: 7281–7287.
- Diamond DM, Bennett MC, Fleshner M, Rose GM (1992) Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2: 421–430. [Crossref]
- Roozendaal B (2002) Stress and memory: opposing effects of glucocorticoids on memory consolidation and memory retrieval. Neurobiol Learn Mem 78: 578–595.
- Woo H, Hong CJ, Jung S, Choe S, Yu S-W (2018) Chronic restraint stress induces hippocampal memory deficits by impairing insulin signaling. Mol Brain 11: [Crossref]
- Korczak DJ, Pereira S, Koulajian K, Matejcek A, Giacca A (2011) Type 1 diabetes mellitus and major depressive disorder: evidence for a biological link. Diabetologia 54: 2483–2493. [Crossref]
- Roy MS, Roy A, Gallucci WT, Collier B, Young K, et al. (1993) The ovine corticotropin-releasing hormone-stimulation test in type I diabetic patients and controls: suggestion of mild chronic hypercortisolism. Metabolism 42: 696–700. [Crossref]
- Magariños AM, McEwen BS (2000) Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci U S A 97: 11056–11061. [Crossref]
- Magariños AM, McEwen BS (1995) Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69: 89–98. [Crossref]
- Magariños AM, Jain K, Blount ED, Reagan L, Smith BH, et al. (2001) Peritoneal implantation of macroencapsulated porcine pancreatic islets in diabetic rats ameliorates severe hyperglycemia and prevents retraction and simplification of hippocampal dendrites. Brain Res 902: 282– 287. [Crossref]
- Zuo Z-F, Wang W, Niu L, Kou Z-Z, Zhu C, et al. (2011) RU486 (mifepristone) ameliorates cognitive dysfunction and reverses the down-regulation of astrocytic N-myc downstream-regulated gene 2 in streptozotocin-induced type-1 diabetic rats. Neuroscience 190: 156–165. [Crossref]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, et al. (2008) Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11: 309–317. [Crossref]
- Stranahan AM, Lee K, Pistell PJ, Nelson CM, Readal N, et al. (2008) Accelerated cognitive aging in diabetic rats is prevented by lowering corticosterone levels. Neurobiol Learn Mem 90: 479–483. [Crossref]
- Sharma VK, Singh TG (2020) Chronic Stress and Diabetes Mellitus: Interwoven Pathologies. Curr Diabetes Rev 16: 546–556. [Crossref]
- Reagan LP, McEwen BS (2002) Diabetes, but not stress, reduces neuronal nitric oxide synthase expression in rat hippocampus: implications for hippocampal synaptic plasticity. Neuroreport 13: 1801–1804. [Crossref]
- Taheri N, Aminorroaya A, Ismail-Beigi F, Amini M (2018) dexamethasone stress test: a pilot clinical study for identification of individuals highly prone to develop type 2 diabetes. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol 24: 894–899.
- Bruehl H, Wolf OT, Sweat V, Tirsi A, Richardson S, Convit A (2009) Modifiers of Cognitive Function and Brain Structure in Middle-Aged and Elderly Individuals with Type 2 Diabetes Mellitus. Brain Res 1280: 186–194. [Crossref]
- Reynolds RM, Strachan MWJ, Labad J, Lee AJ, Frier BM, et al. (2010) Morning cortisol levels and cognitive abilities in people with type 2 diabetes: the Edinburgh type 2 diabetes study. Diabetes Care 33: 714–720. [Crossref]
- Dey A, Hao S, Wosiski-Kuhn M, Stranahan AM (2017) Glucocorticoid-mediated activation of GSK3β promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol Aging 57: 75–83. [Crossref]
- Wamil M, Seckl JR (2007) Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discov Today 12: 504–520. [Crossref]
- Sandeep TC, Yau JLW, MacLullich AMJ, Noble J, Deary IJ, et al. (2004) 11β-Hydroxysteroid dehydrogenase inhibition improves cognitive function in healthy elderly men and type 2 diabetics. Proc Natl Acad Sci U S A 101: 6734–6739. [Crossref]
- de Quervain DJ-F, Poirier R, Wollmer MA, Grimaldi LME, Tsolaki M, et al. (2004) Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum Mol Genet 13: 47–52. [Crossref]
- Barat P, Brossaud J, Lacoste A, Vautier V (2013) Nocturnal activity of 11β-hydroxy steroid dehydrogenase type 1 is increased in type 1 diabetic children. Diabetes Metab 39: 163–168. [Crossref]
- Sooy K, Webster SP, Noble J, Binnie M, Walker BR, et al. (2010) Partial Deficiency or ShortTerm Inhibition of 11 -Hydroxysteroid Dehydrogenase Type 1 Improves Cognitive Function in Aging Mice. J Neurosci 30: 13867–13872. [Crossref]
- Wheelan N, Webster SP, Kenyon CJ, Caughey S, Walker BR, et al. (2015) Short-term inhibition of 11β-hydroxysteroid dehydrogenase type 1 reversibly improves spatial memory but persistently impairs contextual fear memory in aged mice. Neuropharmacology 91: 71–76. [Crossref]
- Dhingra D, Parle M, Kulkarni SK (2004) Memory enhancing activity of Glycyrrhiza glabra in mice. J Ethnopharmacol 91: 361–365. [Crossref]
- Mohler EG, Browman KE, Roderwald VA, Cronin EA, Markosyan S, et al. (2011) Acute Inhibition of 11 -Hydroxysteroid Dehydrogenase Type-1 Improves Memory in Rodent Models of Cognition. J Neurosci 31: 5406–5413. [Crossref]
- Sooy K, Webster SP, Noble J, Binnie M, Walker BR, et al. (2010) Partial Deficiency or ShortTerm Inhibition of 11 -Hydroxysteroid Dehydrogenase Type 1 Improves Cognitive Function in Aging Mice. J Neurosci 30: 13867–13872. [Crossref]
- MacLullich AMJ, Ferguson KJ, Reid LM, Deary IJ, Starr JM, et al. (2012) 11β-hydroxysteroid dehydrogenase type 1, brain atrophy and cognitive decline. Neurobiol Aging 33: 207.e1–8. [Crossref]
- Barat P, Brossaud J, Lacoste A, Vautier V, Nacka F, et al. (2013) Nocturnal activity of 11β-hydroxy steroid dehydrogenase type 1 is increased in type 1 diabetic children. Diabetes Metab 39: 163–168. [Crossref]
- Reul JM, de Kloet ER (1985) Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117: 2505–2511. [Crossref]
- de Kloet ER, Meijer OC, de Nicola AF, de Rijk RH, Joëls M (2018) Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front Neuroendocrinol 49: 124–145. [Crossref]
- Joëls M, Karst H, DeRijk R, de Kloet ER (2008) The coming out of the brain mineralocorticoid receptor. Trends Neurosci 31: 1–7. [Crossref]
- Rozeboom AM, Akil H, Seasholtz AF (2007) Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc Natl Acad Sci U S A 104: 4688–4693. [Crossref]
- Ferguson D, Sapolsky R (2007) Mineralocorticoid receptor overexpression differentially modulates specific phases of spatial and nonspatial memory. J Neurosci Off J Soc Neurosci 27: 8046– 8052. [Crossref]
- Lai M, Horsburgh K, Bae S-E, Carter RN, Stenvers DJ, et al. (2007) Forebrain mineralocorticoid receptor overexpression enhances memory, reduces anxiety and attenuates neuronal loss in cerebral ischaemia. Eur J Neurosci 25: 1832–1842. [Crossref]
- Wingenfeld K, Otte C (2019) Mineralocorticoid receptor function and cognition in health and disease. Psychoneuroendocrinology 105: 25–35. [Crossref]
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M (1998) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19: 269–301. [Crossref]
- Joëls M (2006) Corticosteroid effects in the brain: U-shape it. Trends Pharmacol Sci 27: 244– 250. [Crossref]
- Van Looveren K, Van Boxelaere M, Callaerts-Vegh Z, Libert C (2019) Cognitive dysfunction in mice lacking proper glucocorticoid receptor dimerization. PloS One 14: e0226753. [Crossref]
- de Kloet ER, Oitzl MS, Joëls M (1999) Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci 22: 422–426.
- Oitzl MS, Fluttert M, Sutanto W, de Kloet ER (1998) Continuous blockade of brain glucocorticoid receptors facilitates spatial learning and memory in rats. Eur J Neurosci 10: 3759–3766.
- Revsin Y, Rekers NV, Louwe MC, Saravia FE, De Nicola AF, et al. (2009) Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocin-induced type 1 diabetes mice. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 34: 747–758. [Crossref]
- Plieger T, Felten A, Splittgerber H, Duke É, Reuter M (2018) The role of genetic variation in the glucocorticoid receptor (NR3C1) and mineralocorticoid receptor (NR3C2) in the association between cortisol response and cognition under acute stress. Psychoneuroendocrinology 87: 173–180. [Crossref]
- Caruso A, Nicoletti F, Mango D, Saidi A, Orlando R, et al. (2018) Stress as risk factor for Alzheimer’s disease. Pharmacol Res 132: 130–134. [Crossref]