There is evidence that platelet-derived growth factor (PDGF) is involved in the development of numerous human tumor types. The evidence is most complete in the case of glioblastomas cells. Although many signal transduction pathways have several steps shared with PDGF, we found that inhibition of Jun kinase N-terminal kinase (JNK) stimulate gliomas in an autocrine fashion. Here, we have investigated the role of PDGF in the regulation of human T98G glioblastoma cells grown, in both the presence and absence of a small interfering RNA (siRNA) against Jun-N-Terminal Kinase one (JNK1). Treatment of T98G cells with PDGF alone multiplied substantially faster than untreated cells. In contrast, cells treated with siRNA against JNK-1 led to a reduction in growth rate and DNA synthesis, while cells treated with a combination of PDGF and siRNA failed to alter the growth rate, DNA synthesis and cell proliferation as compared to cells treated only with either, PDGF or siRNA against JNK1 alone. The inhibition of JNK-1 expression by siRNA leads to a marginal inhibition of NF-kB transcriptional activity and strongly reduced the transcriptional activity of the activator protein-1 (AP-1) in presence of PDGF. The reduction was more evident in presence of PMA, a positive control which usually increase the activity of JNK-1 kinase. In conclusion, our results suggest that siRNA against c-Jun N terminal kinase 1 might have an important role in regulating the expression and effect of PDGF in glioblastoma cells.
platelet derived growth factor, T98G, c-Jun-N-terminal kinase (JNK), AP-1
Glioblastoma T98G is a well-studied tumor line which has been reported to express receptors for and proliferate in response to several growth factors including PDGF-like proteins [1], Epidermal Growth Factor (EGF) [2,3] and Insulin-like growth factors (IGF) [4]. Recent studies have revealed that most if not all these factors share common steps in signal transduction. Since PDGF appeared successful as an activator of T98G cells and it is a factor involved in the survival, proliferation, and motility of several cell types we studied the effect of inhibition of c-Jun -N -terminal kinase (JNK-1) by small interfering RNA against JNK-1 in PDGF treated T98G cells. PDGF occurs as homodimers of either the A-chain (AA), B-chain (BB) or as a heterodimer (AB). PR-B preferentially binds the PDGF B-chain whereas PR-A interacts with all three isoforms. Ligand binding promotes receptor dimerization and autophosphorylation [5]. PDGF signaling network consists of four ligands, PDGFA-D, and two receptors, PDGFRa and PDGFRb. All PDGFs function as secreted, disulphide-linked homodimers, but only PDGFA and B can form functional heterodimers [5-10]. Those characteristics of PDGF have been observed in glioma cell lines including cells line T98G as well as for primary gliomas [1,6]. However, the evidence of the effect of PDGF is most complete in the case of glioblastomas which nearly invariably over-express one or both PDGF chains and commonly express one or more receptor subtypes [7-9]. Kinase activation is visualized as tyrosine phosphorylation of the receptor molecules, which occurs between the dimerized receptor molecules [10-12]. Recently, several studies have revealed that PDGF shares with other factors common steps in signal transduction [13,14], such that, ligand-activated receptors bind, phosphorylate, and activate a variety of cytosolic enzymes which may transmit signals. Thus, c-Raf-1 ser/thr kinase activity also has been reported to be increased by PDGF stimulation [15,16]. Two mechanisms may contribute to this result: direct binding to and activation by PR [15,16] and direct phosphorylation by c-Ras [17-19]. Thus c-Raf-1 activation may in turn lead to direct interaction with and activation of MAP Kinase (MAPKK, MEK) [20]. PDGF-activated c-Raf-1 enhances c-Fos transcription which is expected to increase AP-1 complex formation and mitogenesis [20,21]. The c-Ras/c-Raf-1 path also activates AP-1 through phosphorylation of c-Jun. Thus, intervention at a crucial common step may be an important strategy for treating tumors that exhibit multiple autocrine/paracrine mechanisms. One of these steps is the c-Jun-N-Terminal Kinase (JNK) which has been proposed as a crucial target for PDGF [22-24]. The Jun Kinase (JNK) pathway is a member of the mitogen-activated protein kinase (MAPK). The MAPK family can be subdivided into three major groups, i.e., extracellular signal-regulated kinase (Erk), p38, and c-Jun N-terminal kinase (JNK) [24-26]. The JNK is rapidly activated by a large variety of toxic stimuli and inflammatory cytokines suggesting roles it is mediating inflammatory responses, stress responses, and apoptosis [24-26]. Also, previous studies have shown that this pathway participates in the transformation of animal model cells including primary murine fibroblasts [27]. Activation of the JNK pathway leads to the activation of a central kinase which acts directly on three major regulatory proteins, c-Jun, ATF2, and Elk-1 by phosphosphorylating serine residues of activation domains which greatly increases their transcriptional (c-Jun, ATF2) or DNA binding (Elk-l) potential [28]. Similarly, the NF-κB is a family of transcription factors that bind to the enhancer element of the immunoglobulin kappa light-chain of activated B cells [23,24,29,30]. Structurally, NF-κB is composed of homodimers and heterodimers of the 5 members of the Rel family, namely NF-κB1 (p50/p105), NF-κB2 (p52/100), RelA (p65), and c-Rel [29,30,31].
In this study, we showed that treatment of T98G cells with PDGF multiplied substantially faster than naturally growing cells compared to control cells and was strongly affected by the addition of a specific siRNA against JNK-1. The information obtained by the luciferase reporter gene construct demonstrate a prevalence for the AP-1 reporter construct, compared with the NF-kB luciferase construct. In this study, we conclude that JNK-1 is required for growth and PDGF activity in T98G cells and that activation of AP-1 is a common activity of most growth factors that are known to stimulate gliomas.
Cell culture
The T98G brain tumor cell line, established from human glioblastoma was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS, 1 mM sodium pyruvate, 100 Uml-1 penicillin G, 100 mg/ml, 1 streptomycin, 2 mM glutamine, 1 mM MEM non-essential amino acids and 50 mM 2-mercaptoethanol in 5% CO2 incubator at 37°C. The cells were dissociated using 0.25% trypsin and 0.53 mM EDTA solution and subculture once in 3–5 days.
Reagents
Tris-borate-EDTA, TGF-b, and acrylamide: bisacrylamide (29:1) were obtained from Bio-Rad (Richmond, CA, USA). Lipofectamine was obtained from Life Technologies, Inc., USA. Complete Mini-EDTA-Free Protease Inhibitor Cocktail Tablets and Annexin-V-Fluos were purchased from Roche Diagnostics GmbH (Mannheim, Germany). Phorbol 12-myristate 13-acetate (PMA) and TNF-a were purchased from Strategene Inc. (La Jolla, CA, USA). Antibodies JNK (FL): sc-571, JNK‑1 (C-17): sc-474, JNK-2 (N18): sc-827 and β-actin (sc-1616) were used at a concentration of 0.07 and 0.1 mg/ml, respectively (in a total volume of 12 ml), were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA).
DNA synthesis assay
DNA replication rate was measured by a (3H)-Thymidine incorporation assay. Cells were seeded in 96-well tissue culture plates (1,000 cells/well) and treated with PMA (30 ng/ml) or PDGF (100 ng/ml). Twenty-four, 48, 72, 96 and 120 hours after treatment with PMA and /or PDGF (3H)-Thymidine (0.5 mCi/well) was added for 3 hours. Cells were harvested with a PhD-200A cell harvested (Cambridge Technologies, Cambridge, MA), which transferred labeled lysates to paper spots. These were subsequently washed, and the amount of radioactive DNA was quantitated by scintillation counting using Biosafe II scintillation liquid.
Small interfering RNA (siRNA) transfection
T98G glioblastoma cells were cultured to 60-80% confluent on 6-well plates. Cells were incubated with 60 nmol/L siRNAs (Santa Cruz) targeting JNK-1 (sc-29380), JNK-2 (sc-39101) or control “scrambled” siRNA (sc-37007) and 6 μL siRNA Transfection Reagent, according to the manufacturer's recommendation (Santa Cruz) in 1 mL serum-free medium (MEM) at 37°C for 6 hr. After transfection, cells were cultured in complete medium for 42 hrs or until being used. The siRNAs were purchased from Santa Cruz Biotechnology, Inc.
Preparation of cell lysates
T98G human glioblastoma cells were washed once with PBS and suspended in lysis buffer (40 mM HEPES, pH 7.4, with 10% glycerol, 1% Triton X-100, 0.5% Nonidet P-40 (NP-40), 150 mM NaCl, 50 mM NaF, 20 mM β-glycerol phosphate, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulphonyl fluoride and 0.1 mM vanadate) containing a protease inhibitor mixture (1 mg/ml aprotinin, leupeptin and pepstatin). Cell lysates were cleared by centrifugation at 15,000 rpm for 30 min, collected and stored 80˚C.
Transient transfection and AP-1 / NF-kB luciferase assay
Transfections of T98G cells were performed using Lipofectamine TM2000 (Invitrogen) according to the manufacturer’s instructions. The NF-κB reporter plasmid driven by the rat prolactin minimal promoter (-36 to +37) under the control of the two copies of the NF-κB binding site of the human Ig κ light chain enhancer 5'-GGGACTTTCC-3`was kindly provided by M. Rincón and R.A. Flavell (Section of Immunobiology, Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT). The 4xAP-1 reporter plasmid under the control of four copies of a consensus sequence of AP-1 binding site, was create by our laboratory. To assay for luciferase activity, transfected cells in duplicate wells were cultured for 24 h before being stimulated with or without PMA (30 ng/ml) (Positive control) or PDGF (100 ng/ml) for a defined length of time. Cells were harvested, washed twice in PBS and treated with lysis buffer (Luciferase Assay, Promega) for 5-10 min on ice. Lysates were spun down for 1 min, and the total supernatants were analyzed using Luciferase Reagent (Promega) and measured in a luminometer (MicroLumat LB-96P, Berthold) for 5 sec. Background measurement was subtracted from each duplicate, and experimental values are expressed either as recorded light units, luciferase activity or as relative activity compared to extracts from unstimulated cells. Cell lysis and the measurement of reporter luciferase activity were performed by applying the luciferase assay system (Promega, Mannheim, Germany) [32,33].
Western immunoblot analysis
After reaching 70% to 80% confluence, T98G cells were starved overnight with serum-free medium and then treated at the indicated concentrations and periods with different drugs in the absence of serum. Forty-eight hours after transfection, cells were collected and washed twice by cold PBS, and each well was treated with 50 mL lysis buffer (2 mmol/L Tris-HCl pH 7.4, 50 mmol/L NaCl, 25 mmol/L EDTA, 50 mmol/L NaF, 1.5 mmol/L Na3VO4, 1% Triton X-100, 0.1% SDS, supplemented with protease inhibitors 1 mmol/L phenylmethylsulfonylfluoride, 10 mg/L pepstatin, 10 mg/L aprotinin, and 5 mg/L leupeptin) (all from Sigma). Protein concentrations were determined using the Bradford protein assay. Equal amounts of protein (40 mg) were separated on a 15% SDS polyacrylamide gel and transferred to a nitrocellulose membrane (Hybond C, Amersham, Freiburg, Germany). Membranes were blocked in 5% nonfat dry milk in TBS for 1 h at room temperature and probed with rabbit antiEgr-1 antibodies (dilution, 1:500 Santa Cruz Biotechnology, USA) overnight at 4ºC. After 3 times washing with TBS containing 0.1% Tween 20, membranes were incubated with anti-rabbit IgG-horseradish-peroxidase (1:5000, Santa Cruz Biotechnology, USA), and developed by luminol mediated chemiluminescence (Appylgen Technologies Inc, China). To confirm equal protein loading, membranes were reprobed with a 1:1000 dilution of an anti-actin antibody (Santa Cruz Biotechnology, USA). Densitometric analyses were performed using Scion Image software.
Growth and viability assays
Cell viability was analyzed by the determination of the viable cell mass by the addition of 3-(4,5- dimethylthiazol-2-y1-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt, (MTS) in 96-well cluster plates in accordance with the manufacturer's protocol (Promega, Madison, WI, USA). All viability determination assays were carried out in triplicate and cell viability was expressed as the ratio of viable cell mass following a given treatment relative to that of parallel cultures of untreated cells x 100 (Cell Viability, %).
The effect of PDGF in cells is well documented and therefore we thought that the T98G cells were an appropriate choice to study the effect of PDGF, because their well characterized growth properties, lack of p53, and a detectable basal level of JNK activity.
Expression of PDGF and JNK-1 protein in human T98G glioblastoma cell line
The N-terminal Jun Kinase/Stress-Activated Protein Kinase (JNK/SAPK) signal transduction is rapidly activated by a large variety of toxic stimuli and inflammatory cytokines suggesting roles in mediating inflammatory responses, stress responses, and apoptosis. To determine the effect of blocking the expression of the kinase JNK-1 by small interfering RNA, and its effect on the activity of PDGF, we first studied the protein expression of genes encoding the JNK-1 and PDGF and its products in T98G cells. The cells were treated with several inductor of JNKs, such as, with FCS 0,5%, FCS 10%, TGF-b1 (10 ng/ml), PMA (30 ng/ml) and TNF-a (20 ng/ml). As showed in Figure 1, the protein expression of JNK-1 was assessed by western blot analysis (Figure 1). The basal protein levels of both, JNK1 was higher in cells cultured in FBS 10%, compared to cells cultured in FBS 0.5%. Treatment with PMA and TNF-a strongly activated JNK in all cases.
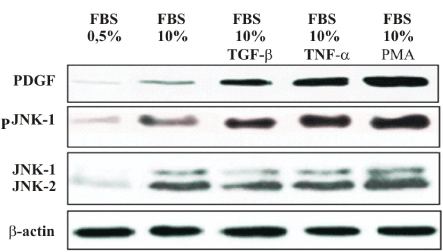
Figure 1. JNK-1 and PDGF expression in T98G glioblastoma cell lines. To determine activates the Jun kinase (JNK) pathway in LNCaP and PC-3 cells, the cells were grown in 0.5 and 10% FBS and treated either with TGF-b (300 µg/ml) or PMA (30 ng/ml) or TNF-a (30 ng/ml 30 min). PDGF and JNK-1 protein expression were examined 2 h after treatment, by western blot. One of three similar experiments is shown
Expression of the small interfering RNA against JNK-1 causes inhibition of activated AP-1 and NF-kB mediated transactivation
The studies with siRNA-JNK in prostate cell lines, suggested that AP-1 activity may be involved in transformation by v-sis. We, therefore, examined the activation of AP-1 and NF-kB by transfection of the T98G model cells with a pGL-2- reporter constructs containing an AP-1 and/or a NF-kB sensitive promoter with the consensus sequence of response elements of the above mentioned nuclear factors. In order to test whether siRNA-JNK-1 is an effective inhibitor of the kinase JNK-1 and thereby inhibiting the transcriptional activity of nuclear factors AP-1 and NF-kB, transient cotransfection assays were performed. Two Luciferase-based reporter constructs regulated by binding of AP-1 and NF-kB transcription complexes to multiples copies of the consensus sequence of AP-1 (pGL-2-4xAP-1) and consensus sequence of NF-kB (pGL-2-2xNF-kB) site were examined. The cells transfected with the reporter constructs were treated with PDGF (100 ng /ml) and PMA (30 ng/ml). Scrambled siRNA was used to examine the specificity of inhibition by siRNA-JNK-1. Thus, specific inhibition of the activated JNK-1 pathway is predicted to strongly inhibit the AP-1 but not NF-kB dependent reporters. The two reporters (AP-1 and NF-kB) treated with PMA or PDGF showed enhanced expression (between 3 to 10-fold in T98G cells). We then cotransfected the siRNA-JNK-1 inhibitor of JNK-1 kinase. As controls, parallel transfection was carried out with a scrambled siRNA. Expression of siRNA-JNK-1, but not scrambled siRNA, led to substantial inhibition (60 %) of the AP-1 reporter activity, whereas inhibition of the NF-kB reporters was much lower. The observation that expression of siRNA-JNK-1 strongly inhibits PMA and PDGF effect in 4xAP-1-reporter constructs is consistent with the role of N-terminal phosphorylation of c-Jun in the AP-1 sequence-dependent systems in cells. Therefore, expression of the siRNA-JNK-1 in T98G cells is expected to strongly inhibit the AP-1 regulated genes in cells with an activated JNK pathway. The fact that the inhibition of NF-kB reporter construct was weaker that the response of AP-1-Luc, suggest that the AP-1 inhibition may involve a direct effect on JNK kinase (Figures 2 and 3).
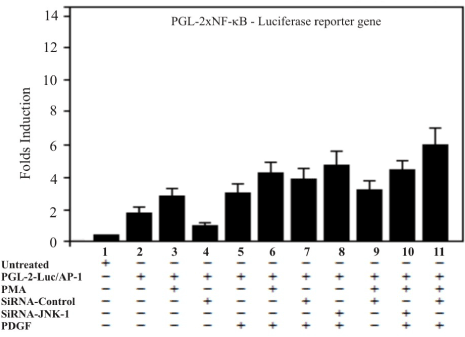
Figure 2. Treatment of cells with PDGF potentiates thetranscription activityof NF-kB luciferase reporter gene but only marginally is affected by JNK-1-siRNA. Twenty-four hours after the transfection T98G cells were treated with siRNA against JNK-1 and activated with PMA and PDGF. The luciferase reporter carrying two NF-kB response elements in tandem directing expression was transfected into T98G cells. The graph shows NF-kB luciferase activity compared to the controls with a reporter lacking the NF-kB site. A representative of three independent experiments carried out in triplicates is shown. A representative of three independent experiments carried out in triplicates is shown. Error bars indicate the S.D. of three determinations
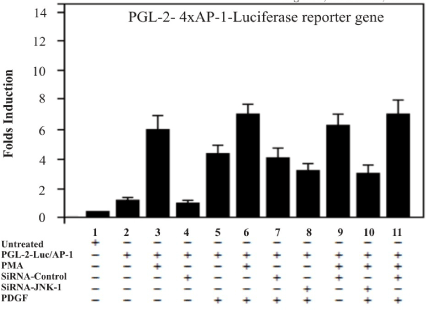
Figure 3. AP-1 transcriptional activity is inhibited by expression of JNK-1-siRNA. Inhibition of AP-1-dependent transcription induced by PMA stimulation was compared with the effects of the small interfering siRNA against JNK-1 (JNK-1-siRNA). The reporter, which contains 4xAP-1 response elements in tandem directing expression of the luciferase gene, was transfected into T98G cells. The graph shows luciferase activity compared to the controls with a reporter lacking the AP-1 sites. The AP-1 reporter requires an activated JNK-1 kinase and was therefore affect by the expression of the siRNA-JNK-1. A representative of three independent experiments carried out in triplicates is shown. Error bars indicate the S.D. of three determinations
Growth inhibition in T98G cells by the small interfering RNA against JNK-1 kinase
To determine whether elimination of JNK-1 has any effect on the proliferation rate induced by PDGF and PMA of T98G cells, we carried out proliferation assays following lipofection with siRNA against JNK-1 and control scrambled siRNA. In addition to transfection of cells with siRNA-JNK-1, the T98G cells were treated with two well-known JNK-1 kinase inducers, the PMA (30 ng/ml) and PDGF (100 ng/ml).
Treated cells were allowed grow for 48 hours after the lipofection. Cells treated with scrambled siRNA displayed growth characteristics like de untreated control cells (parental) (Figure 4, lane 1, lane 2). T98G cells treated with either, PMA (30 ng/ml) or PDGF (100 ng/ml) exhibited a strong increment in cell growth (Figure 4, lanes 3, 5, 6, 8, 9, 11). In contrast, cells treated with siRNA-JNK-1 exhibited a marked reduction in cell growth (Figure 4, lane 4, 7, 10, 12). The proliferation of siRNA-JNK-1-treated cells was approximately half of that of control cells, and approximately 60 % or more compared with cells treated with PMA or PDGF (Figure 4, lane 3, 5, 6, 9, 11).
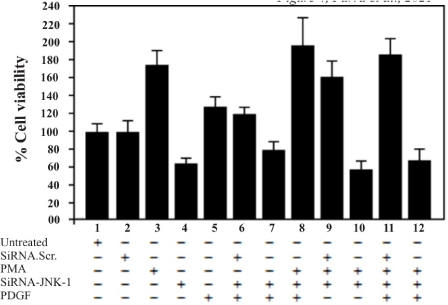
Figure 4. T98G cells were treated with either, PDGF or PMA and seeded in 96-well tissue culture plates at density of 5x103 cell/well and lipofected with 0.4 mM of JNK-1-siRNA and scrambled of control siRNA next day. Cells were stained with MTS/PMS and the protein was determined by measurement of optical density at 490 nm as described (Materials and Methods). A representative of three independent experiments carried out in triplicates is shown. Error bars indicate the S.D. of three determinations
Blockage of JNK-1 expression by small interfering RNA inhibits basal growth of T98G cells and reduces the positive effect of PMA and PDGF treatment
To test the role of the JNK-1 pathway on cell growth, T98G cells were transfected with siRNA against JNK-1. The proliferation rates of untreated T98G cells, and five groups of either, treated, transfected or both cells were examined over the course of ten days (Figure 5). Parental T98G cells grew to high densities of approximately 280×103 cells/cm2. In contrast, cells treated with PMA or PDGF exhibited an increase of both, proliferation rate and saturation density. In contrast, cells treated with siRNA-JNK-1 exhibited a marked in both proliferation rate and saturation density.
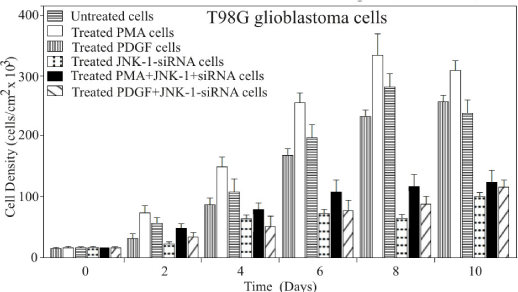
Figure 5. T98G cells dosed with PMA or PDGF proliferate at expected rates with respect to the control cells. T98G cells were plated in multiwell tissue culture plates at 25x103 cells in medium containing 10% FCS and counted (Coulter counter) on the indicated days in triplicate. The average values are plotted here to define the growth of the cells. The maximum proliferation is plotted as a function of PMA and PDGF to increase cell growth or proliferation. The minimum proliferation of the cells is plotted as a function of JNK-1 -siRNA to inhibit or delayed cell growth. A representative of three independent experiments carried out in triplicates is shown. Error bars indicate the S.D. of three determinations
Glioblastoma T98G cell is a well-studied tumor line which has been reported to express receptors for and proliferate in response to several growth factors including PDGF-like proteins [34], EGF [35] and FGF [36]. Several studies have also revealed that glioblastomas invariably over-express one or both PDGF chains and commonly express one or more receptor subtypes. Moreover, the PDGF signal transduction pathway has several steps shared by other growth factors, some of which are known to stimulate gliomas in an autocrine fashion [36]. These common steps are, therefore, attractive targets for intervention and one of them is the Jun-N-Terminal kinase pathway, a member of the MAP kinase family. There is evidence that phosphorylation by PDGF receptors (PR) causes activation of the recruited enzymes. Thus, PLCg has been reported to be activated by PR [37,38] leading to increased hydrolysis of phosphoinositides (Pl). Pl metabolism leads to diacylglycerol (DAG) and calcium mobilization, both of which activate protein kinase-C (PKC) which in turn leads to activation of c-Jun [37,38]. On the other hands, c-Raf-1 activation may in turn lead to direct interaction with and activation of MAP Kinase (MAPKK, MEK) [39,40]. PDGF-activated c-Raf-1 enhances c-Fos transcription which is expected to increase AP-1 complex formation and mitogenesis [39,40]. The goal of this study was to investigate the effects of silencing JNK-1 gene expression with specific small interfering RNA (siRNA) on PDGF activity in T98G cells. In this way, to understand the molecular effects of PDGF on T98G cells and its relationship to the JNK-1 kinase pathway, we examined the effect of this factor on the regulation of JNK-1 and the transcriptional activity of two nuclear factors, namely, AP-1 and NF-kB. We observed that inhibition of JNK by a specific siRNA prevented both, the kinase activity of JNK-1 (probably by blocking the phosphorylation of c Jun) and the induction of AP-1 transcriptional activity. We, therefore, examined the activation of AP-1 using a luciferase construct containing four sensitive AP-1- response elements (pGL-2x4AP-1), compared with a luciferase construct containing two NF-kB response elements (pGL-2x2NF-kB) [31-33]. Our observation indicates a regulation function that involved JNK-1, suggesting that activation of AP-1 is a common activity of most if not all growth factors that are known to stimulate human gliomas [34-36], but at the same time, the results reveal PDGF as a pivotal component of the glioblastoma cell tumorigenic, sharing this activity with different signaling pathways such as the Jun N terminal kinase. The conclusion is that addition of siRNA-JNK-1, which has no effect on control cells, strongly decrease cell proliferation (Figure 5) and cell viability (Figure 4). Similarly, cotransfection of siRNA-JNK-1 with either pGL-2x4-AP-1 or pGL-2x2-NF-kB, strongly decrease AP-1 luciferase activity and moderately the activity of the pGL2-x2kB. PMA or PDGF had the expected effects and served as positive controls while an empty plasmid served as negative controls. Indeed, it is well known that c-Jun transcriptional activity is regulated by JNK-mediated phosphorylation of the NH2-terminal transactivation domain on Ser63 and Ser73 [41,42]. Thus, the observation that inhibition of JNK-1 gene expression by a specific siRNA against JNK-1 resulted in a significant reduction of PDGF and JNK-1 levels, provided the evidence that JNK-1 plays a dominant role in mediating growth and survival of T98G cells in response to PDGF, and that effect may be partially attributable to AP-1 activation.
We thank Dra. M. Rincon and R.A. Flavell [Section of Immunobiology, Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT] for providing the reporter gen vector carrying the 2x NF-κB consensus sequence. This work was supported by grants from Universidad de Tarapacá. Project-UTA-Mayor-7701-2018. The authors declare that they have no competing interests.
- Pollack IF, Randall MS, Kristofik MP, Kelly RH, Selker RG, et al. (1990) Response of malignant glioma cell lines to epidermal growth factor and platelet-derived growth factor in a serum-free medium. J Neurosurg 73: 106-112. [Crossref]
- Takahashi JA, Fukumoto M, Kozai Y, Ito N, Oda Y (1991) Inhibition of cell growth and tumorigenesis of human glioblastoma cells by a neutralizing antibody against human basic fibroblast growth factor. FEBS Lett 288: 65-71. [Crossref]
- Píate KH, Breier G, Farrell CL, Risau W (1992) Platelet-derived growth factor receptor-beta is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab Invest 67: 529-534. [Crossref]
- Ruslan Novosyadly, Dudas J, Pannem R, Ramadori G, Scharf JG (2006) Crosstalk between PDGF and IGF-I receptors in rat liver myofibroblasts: implication for liver fibrogenesis. Lab Invest 86: 710-723. [Crossref]
- Fredriksson L, Li H, Eriksson U (2004) The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev 15: 197-204. [Crossref]
- Nister A, Libermann T, Betsholtz C, Pettersson M, Claesson-Welsh L, et al. (1988) Expression of mRNAs for PDGF and transforming growth factor-a and their receptors in human malignant glioma cell lines. Cancer Res 48: 3910-3918. [Crossref]
- Liu KW, Hu B, Cheng SY (2011) Platelet-derived growth factor signaling in human malignancies. Chin J Cancer 30: 581-584. [Crossref]
- Huang F, Wang D, Yao L, Wang M (2017) PDGF signaling in cancer progression. Int J Clin Exp Med 10: 9918-9929.
- Mauro A, Bulfone A, Turco E, Schiffer D (1991) Coexpression of platelet-derived growth factor (PDGF) B chain and PDGF B-type receptor in human gliomas. Childs Nerv Sys 7: 432-436. [Crossref]
- Lokker N, Sullivan CM, Hollenbach S, Israel M, Giese N (2002) Platelet-derived Growth Factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res 62: 3729-35. [Crossref]
- Baxter RM,Secrist JP,Vaillancourt RR,Kazlauskas A (1998) Full activation of the platelet-derived growth factor beta-receptor kinase involves multiple events.J Biol Chem 273: 17050-17055. [Crossref]
- Heldin CH (2013) Targeting the PDGF signaling pathway in tumor treatment.Cell Commun Signal 11: 97-98. [Crossref]
- Elizabeth SM, Andrea DH, Ryaz BC, Lindsey MJ, Deborah HA (2005) A-Raf associates with and regulates platelet-derived growth factor receptor signaling. Cell Signal 17: 857-868. [Crossref]
- Erxi Wu, Nathan P, Ze T, Annie PM, Michal G, et al. (2008) Kohane comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS One 3: e3794. [Crossref]
- Jamal S, Ziff E (1990) Transactivation of c-fos and beta-actin genes by raf as a step in early response to transmembrane signals. Nature 344: 463-466. [Crossref]
- Kaplan DR, Escobedo JA, Rapp UR, Roberts TM, Williams LT, et al. (1989). Direct activation of the serine/threonine kinase activity of raf-1 through tyrosine phosphorylation by the PDGF B-receptor. Cell 58: 649-657. [Crossref]
- Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M (1993) Complex formation between RAS and RAF and other protein kinases. Proc Nati Acad Sci USA 90: 6213-6217. [Crossref]
- Vojtek A, Hollenberg S, Cooper J (1993) Mammalian ras interacts directly with the serine/threonine kinase Raf. Cell 74: 205-214. [Crossref]
- Wu H (2013) Higher-Order assemblies in a new paradigm of signal transduction. Cell 153: 287-292. [Crossref]
- Jamal S, Ziff E (1990) Transactivation of c-fos and beta-actin genes by raf as a step in early response to transmembrane signals. Nature 344: 463-466. [Crossref]
- Kaibuchi K, Fukumoto Y, Oku N, Hori Y, Yamamoto T, et al. (1989) Activation of the serum response element and 2-0-tetradecanoylphorbol-13-acetate response element by the activated c-raf-1 protein in a manner independent of protein kinase C. J Biol Chem 264: 20855-20858. [Crossref]
- Yu J, Deuel TF, Kim HR (2000) Platelet-derived Growth Factor (PDGF) Receptor-Alpha Activates c-Jun NH2-terminal kinase-1 and Antagonizes PDGF Receptor-Beta -Induced Phenotypic Transformation. J Biol Chem 275: 19076-19082.
- Goldberg HJ, Huszár T, Mózes MM, Rosivall L, Mucsi I (2002) Overexpression of the type II transforming growth factor-beta receptor inhibits fibroblasts proliferation and activates extracellular signal regulated kinase and c-Jun N-terminal kinase. Cell Biol Int 26: 165-174. [Crossref]
- Parra E (2012) Inhibition of JNK-1 by small interference RNA induces Apoptotic Signaling in PC-3 Prostate Cancer Cells. Int J Mol Med 30: 923-930. [Crossref]
- Johnson GL, Nakamura K (2007) The c-Jun kinase/stress-activated pathway: Regulation, function, and role in human disease. Biochim Biophys Acta 1773: 1341-1348. [Crossref]
- Rincon M, Davis RJ (2009) Regulation of the immune response by stress-activated protein kinases. Immunol Rev 228: 212-224.
- Johnson R, Spiegelman B, Hanahan D, Wisdom R (1996) Cellular transformation, and malignancy induced by ras require c-jun. Mol Cell Biol 16: 4504-4511. [Crossref]
- Bogoyevitch MA, Kobe B (2006) Uses for JNK: The many and varied substrates of the c-Jun N-Terminal kinases. Microbiol Mol Biol Rev 70: 1061-1095. [Crossref]
- Giuliani C, Bucci I, Napolitano G (2018) The role of the transcription factor nuclear factor-kappa b in thyroid autoimmunity and cancer. Front Endocrinol 9: 471- 478. [Crossref]
- Parra E, Varga M, Sigvardsson M, Hedlund G, Kalland T, et al. (1995) Costimulation of CD4 cells with B7 and LFA-3 induce distinct effects on AP-1 and NF-kB. J Immunol 155: 1132-1141. [Crossref]
- Parra E, McGuire K, Hedlund G, Mikael Dohlsten (1998) Overexpression of Rel A and c-Jun substitutes for B7-1 costimulation by targeting the CD28RE within the IL-2 promoter. J Immunol 160: 5374-5381. [Crossref]
- Parra E, Ferreira J, Ortega A (2011) Overexpression of EGR-1 modulates the activity of NF-kB and AP-1 in prostate carcinoma PC-3 and LNCaP cell lines. Int J Oncol 39: 345-352. [Crossref]
- Parra E, Ferreira J, Saenz L (2011) Inhibition of Egr-1 by siRNA in prostate carcinoma cell lines is associated with decreased expression of AP-1 and NF-kB. Int J Mol Med 28: 847-853. [Crossref]
- Makarenko I, Hede SM, He X, Hedrén A, Thompson J, et al. (2012) PDGF and PDGF receptors in glioma. Ups J Med Sci 117: 99 - 112. [Crossref]
- Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, et al. (1994) Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene 9: 2313-2320. [Crossref]
- Westphal M, Brunken M, Rohde E, Herrmann HD (1988) Growth factors in cultured human glioma cells: Differential effects of FGF, EGF and PDGF. Cancer Lett 38: 283-296. [Crossref]
- Coughlin SR, Escobedo JA, Williams LT (1989) Role of phosphatidylinositol kinase in PDGF receptor signal transduction. Science 243: c1191-1194. [Crossref]
- Moodie SA, Willumsen BM, Weber MJ, Wolfman A (1993) Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase. Science 260: 1658-1661. [Crossref]
- Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179-185. [Crossref]
- Rapp UR, Goldsborough MD, Mark GE, Bonner TL, Groffen J, et al. (1983) Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci USA 80: 4218-4222. [Crossref]
- Kallunki T, Deng T, Hibi M, Karin M (1996) c-Jun Can Recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87: 929-939. [Crossref]
- Atsaves V, George V, Rassidakis Z, Claret F (2019) AP-1 Transcription factors as regulators of immune responses in cancer. Cancers (Basel) 11: 1-21. [Crossref]