Had we but world enough, and time,
This coyness, Lady, were no crime.
We would sit down and think which way
To walk and pass our long love’s day …
The grave’s a fine and private place,
But none, I think, do there embrace.
Now therefore, while the youthful hue
Sits on thy skin like morning dew,
Rather at once our time devour
Than languish in his slow-chapt power [1] ….”
DNA is a coy mistress, difficult to interpret, devouring of too much time for busy practitioners. Yet technology’s power in the form of genomic testing [2,3](Table 1) is now so all-encompassing that no caring physician would let their families languish in molecular confusion. Genomics can postpone the grave’s embrace, its pivotal impact on reproduction and pregnancy justifying a feminine metaphor.
Table 1. Guide to genetic testing.
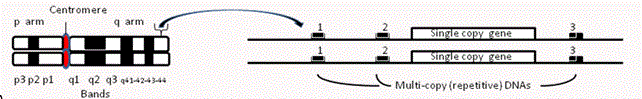
Genome, chromosomes, genes--DNA in the form of 23 chromosome pairs (DNA chains) and their 23,000 component genes (links on the chain) comprises the human genome, established by gamete fusion as the fertilized egg. This founding nuclear genome, complemented by a thousand mitochondria from egg cytoplasm, is copied to make a trillion adult cells comprising >230 tissues. Each tissue, aside from cancer or somatic mutation, contains the same 3-billion base pair genome sequence. NextGen/massive parallel sequencing techniques [14] accelerated DNA sequencing from hundreds to millions of nucleotides per day and specified the sequence of every human chromosome within our gene collection through the Human Genome Project [15]. Composing a consensus text for the human genome [now in its 19th edition with few gaps-16] allowed paging through the 23 chromosome chapters for altered DNA dosage or nucleotide sequence via whole exome sequencing [WES—2]—see below). Our nuclear genome consists of protein-coding (gene) islands amidst multi-copy repetitive DNAs of known versus unknown functions (ribosomal, transfer, microRNA genes versus interspersed, clustered, or mobile repetitive DNA sequences). Diagram A below shows paired chromosomes (all but X-Y in males) with band numbers to reference chromosome regions--a small region is expanded to show paired gene and surrounding repetitive DNAs 1-3, each with similar DNA sequence.
A
|
|
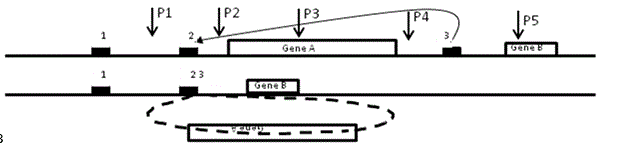
Karyotype to microarray--genetic testing began with microscopic examination of chromosomes (karyotype) to define extra (duplicated) or missing (deleted) chromosomes or chromosome regions (aneuploidy) as found in Down, Turner, or Cri-du-chat syndromes. These errors of meiotic division were complemented by smaller copy number variants (CNVs) generated by pairing of non-allelic repetitive DNAs (e.g., 2-3 rather than 2-2 in diagram B below). After recombination that is ubiquitous among chromosome partners, the unpaired DNA on one chromosome loops out to produce a gene-containing microdeletion (illustrated below) or doubles itself (not shown) to duplicate the gene and its surrounding DNA. Probes P2-P4 (see microarray/WES below) would signal less DNA compared to a normal control while that signaled by probes P1 and P5 would be equal.
B
|
|
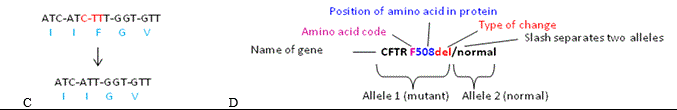
Gene mutations (DNA sequence variants)--microarray or WES scanning for extra or missing DNA segments (copy number variants or CNVs) is analogous to the bookseller who checks the genome book for extra/missing pages, while whole exome or whole genome sequencing is analogous to the proofreader/editor who checks the entire book text for typos. Typos of DNA sequence involve smaller changes than those detected by microarray, including substitution, deletion, or insertion of one or a few DNA base pair letters. The great advance was ability to sequence all DNA regions simultaneously (massive parallel or NextGen DNA sequencing), using the same millions of probes as in microarray but this time extending their ends using a DNA polymerase. The order in which differently colored fluorescent nucleotides were added reflected the order of DNA nucleotides, iterated from all probes to give complete DNA sequences. Comparison of patient to reference gene sequences showed mutations like the missing CTT base pairs (diagram C) that cause the common deltaF508 cystic fibrosis mutation in Caucasians [this mutation ablates a phenylalanine--F in amino acid code—to alter CFTR protein function—17]. Diagram D shows the usual terminology of DNA sequencing reports, specifying normal and delF508 alleles that diagnose the individual as a CF carrier.
|
|
Types of DNA sequencing tests--Probes can target one gene (targeted DNA tests for Marfan syndrome or neurofibromatosis), a group of genes causing related symptoms (DNA testing panels for spinal muscular atrophy or epilepsy), or all protein-coding/exon regions (WES). WES optimally includes parental samples to help determine which of the 12,000 DNA variants in an average individual match up with individual symptoms. Most genes are sequenced at least 10 times (coverage) to ensure accuracy, but the WES technique can focus on particular genes that may relate to patient symptoms or cause unrelated diseases (“secondary or incidental findings”) like the breast–ovarian cancer or BRCA gene mutations [18] that require separate patient consent to be reported. Repeated sequencing of the ~45 million protein-coding (exonic) DNA sequences by WES (~1.5% of the genome) is much more comprehensive and informative than the multiple single nucleotide polymorphisms or SNPs marketed to consumers for ancestry or disease association for costs as low as $100. Single mutation tests cost from $350 to $500, gene panels $500 to $1000, and WES $2000 to $2500 depending on the laboratory. Insurance coverage is increasing with half of my patients (GW) having zero out-of-pocket costs. |
While many phases of genomic testing [4](Table 2) remain in their youthful hue it is particularly necessary that the doctor code walk DNA tests from rare disease to population screen. Genomics can reach every family and define any genetically influenced disease, so it must be embraced by medicine at large and not remain a private place for ward-deprived genetic counselors and lab-contained geneticists. The tables and case examples should remind physicians that 2D DNA becomes coy and complex only when correlated with 4D disease, the essence of general medicine. Physician experience will be just as necessary to place the many results from DNA analysis into patient context as it is to interpret those of a sequential multi-channel analysis with computer-20 (SMA20) panel.
Table 2. Six phases of genetic testing.
Phase |
Types of genetic testing |
Preconception |
Couple with infertility
Couple with recurrent pregnancy loss |
Past: Routine karyotype on couple following infertility evaluation; karyotyping of abortus material
Present: Microarray for Y chromosome deletion [19] causing oligospermia or for subtle aberrations in abortus material
Future: Genome scanninga detects all chromosome aberrations in couples or abortus material, finds novel genes impacting conception/early embryogenesis; ART/IVF and PGD enhance fertility, exclude harmful mutant alleles |
Couples at risk for particular genetic disease (family history, prior affected child)
Low-risk couples desiring preconception screening |
Past: Specific mutant allele testing couples at risk based on their family history or ethnicity (e. g., CF).
Present: Targeted DNA testing available for most chromosome or single gene disorders; preconception screening of >200 genes known to cause recessive disorders; invasive prenatal diagnosisb or costly PGD available for fetal mutant alleles.
Future: Preconception genome scanning for all couples with focus on at-risk mutant alleles followed by options for fetal detection by PGD, testing fetal cell-free DNA from maternal blood [NIPT-20], later prenatal diagnosis.c |
Pregnancy |
Fetuses at risk for specific genetic disease
(family history, prior affected child)
Fetuses at risk due to parental age or birth defect(s) detected by ultrasound
Fetal genetic screening |
Past: At-risk couples have prenatal diagnosis for limited number of chromosomal or single gene diseases. Fetal screening by ultrasound/maternal serum marker levels (triple test, quad screen) not helpful for most genetic disorders.
Present: Prenatal diagnosis expanded to most at-risk couples as many single gene (Mendelian) diseases are characterized at the DNA level. NIPT detects common trisomies/monosomies.
Future: Most carriers of mutant alleles or chromosome changes are identified by preconception genome scanning allowing options for PGD, NIPT (detecting gene as well as chromosome change), or targeted prenatal diagnosis. Fetal genomic scanning in the first/second (NIPT, CVS) or third trimester3 to enhance reproductive/management options |
Newborn/Early Infantile |
Neonate with multiple defects, likely syndrome
Neonate with predominant single birth defect
Newborn with suspect inborn metabolic error
Newborn screening. |
Past: Routine karyotype for newborns with birth defects, sometimes accompanied by a targeted FISH test based on clinical symptoms. Different testing approach for inborn errors and newborn screening by quantifying blood and urine metabolites or qualitative bacterial inhibition/marker detection (e. g., phenylalanine, hormones, hemoglobin)
Present: Microarray demonstrating any type of chromosome change, expanded metabolic screening; specific gene or gene panel testing based on type of birth defect or metabolic testing results.
Future: Genome scanning supplementing the newborn metabolic screen with detection of any chromosome imbalance or mutant alleles associated with developmental, birth defect, metabolic, hormonal or cellular defects (e.g., SCID). |
Childhood—medical disorders |
Children with neurobehavioral disorders
Children with other medical conditions
|
Past: Routine karyotype low (2-3%) yield for etiology of ID/autism without birth defects or dysmorphology. Other conditions might provoke protein and DNA tests for particular genetic diseases (e. g., CF, alpha-1-antitrypsin deficiency, Hurler syndrome); yields depended on appropriate diagnostic suspicion.
Present: Microarray analysis higher (~20%) yield for ID/autism; DNA sequencing panels target genes implicated in ID/ autism. Gene panels also available for other conditions like epilepsy, arrhythmia, cardiomyopathy; WES often used for unusual presentations or rare disorders.
Future: Genome scanning to supplement prior preconception and newborn screening that may miss or misinterpret mutant alleles. Physician guidance necessary to correlate symptoms with mutant alleles and improve screening accuracy. |
Childhood—susceptibilities |
Infants for SIDS, immune/allergic susceptibility
Children for cancer susceptibility
Children destined for procedures or medications
Teens for sports risks and injuries
|
Past and present: Skin testing for allergic diathesis, modification of vaccination schedules after side effects, mostly after symptoms or reactions have occurred. Cancer testing dependent on positive family history or associated clinical changes (e. g., multiple nevi, hemihypertrophy, syndromes like Beckwith-Wiedemann). Uniform procedures (radiation dose, pre-operative medications) not tailored to genetic susceptibility. Sports physicals have low sensitivity for cardiac or skeletal susceptibilities unless expensive sub-specialty evaluations are recommended.
Future: Pre-symptomatic screening using gene panels or WES focused on genes relevant to immune/allergic response, inherited cancer, or cardiac and connective tissue disease; Genome scanning to explain unusual symptoms or diagnosis after SIDS/SUDS or other untoward events. |
Adults—medical disorders and susceptibilities |
Adults with medical conditions including mental illness and other multifactorial disorders
Adult disease susceptibilities and medication sensitivities (precision medicine, pharmacogenomics)
Reproductive genetics returns to the preconception phase as the next generation is planned |
Past: Few DNA tests were available for adult medical conditions, heralded by pre-symptomatic testing for the expanded triplet repeats causing Huntington chorea. Testing for multifactorial disorders (multiple genes plus environmental factors) employed non-DNA biologic markers like lipid profiles for heart disease, HLA B27 for ankylosing spondylitis
Present: Microarray aberrations are similar in schizophrenia and autism, DNA or gene panel testing available for diseases like hemochromatosis, Marfan syndrome, or early onset breast-ovarian cancer. Cytochrome genotypes guide medication dosage and common DNA variations (e.g., MTHFR C677T and A1298C) are over-emphasized in functional medicine.
Future: Genome-wide screening will unify disease and susceptibility testing, posing new challenges but dramatizing the old truth that physicians are best qualified to integrate laboratory testing with patient and family care. |
aGenomic scanning can now be done as a single test that looks for extra/missing chromosome/DNA material (microarray analysis) and whole exome sequencing (WES) for gene changes (mutant alleles); bchorionic villus sampling (CVS) at 10-12 weeks, late first trimester, or amniocentesis at 14-16 weeks, second trimester; cthird trimester amniocentesis or cordocentesis; ART, artificial reproductive technology including in vitro fertilization (IVF) to promote fertility or to produce embryos for preimplantation genetic diagnosis (PGD); CF, cystic fibrosis; CVS, chorionic villus sampling; FISH, fluorescent in situ hybridization; MTHFR, methylene tetrahydrofolate reductase; C677T, A1298C, nomenclature for mutant alleles, indicating cytidine to thymidine substitution at gene position 677 or adenosine to cytidine substation at position 1298; NIPT, non-invasive prenatal testing using cell-free fetal DNA in maternal blood; PGD, preimplantation genetic diagnosis; SCID, severe combined immunodeficiency; SIDS or SUDS—sudden infant or sudden unexplained death syndromes.
DNA is the common denominator unifying previously separate testing categories like karyotyping for children with birth defects, blood/urine chemistries for suspected metabolic disease, or specific subspecialty tests like those for cystic fibrosis in pulmonology/perinatology, spinal muscular atrophy in neurology, or for the Philadelphia chromosome in oncology [2]. Now general physicians can initiate testing for these various disease categories because all can be diagnosed by the same genome-scanning test: whole exome sequencing (WES) that looks simultaneously for extra/missing chromosome segments (formerly done by karyotyping or microarray analysis) and for “typos” in the DNA nucleotide GATTACA sequence that makes its encoded protein dysfunctional (see Table 1). Subspecialists will be involved in the testing for rarer diseases and categories, but family physicians are invaluable for deciding when and which types of testing/subspecialty evaluations are best for families. Even more important is their translation of results into the prevention and monitoring strategies of a medical home.
The first genetic tests began with a suspect diagnosis, confirmed by demonstrating altered structures of targeted genes or their protein products. Children with likely Down syndrome had extra chromosome 21 DNA by karyotype and then microarray, those with anemia and crises had abnormal hemoglobin S, those with intellectual disability and mousy smell had elevated plasma phenylalanine. Newborn screening included the latter two disorders and prenatal sampling of fetal cells allowed testing for all three disorders once DNA made any tissue suitable for testing.
Array formats and NextGen techniques accelerated DNA sequencing from thousands to millions of nucleotides per day, allowing screening of all 23 chromosome pairs for abnormal dosage and most of the 23,000 genes for abnormal nucleotide sequence (Table 1). Six phases of genetic testing can be outlined (Table 2), all of them potentially involving new approaches to medical testing: Screening that avoids the presumption of diagnosis and pre-symptomatic diagnosis that transcends the presumption of disease.
It is the general physician who strives to know the general world of medicine, and their time, though limited, can make a huge difference for families as shown by the following case examples. Supporting this valuable service are accessible web resources that offer describe clinical symptoms and DNA changes for any genetic disease [5], outline symptom patterns for particular DNA dosage changes/chromosome disorders [6], give details on genes and their DNA sequence changes [7] for those seeking more detail than laboratory reports provide.
A 3-year-old boy had mild motor but significant speech delay along with sensitivity to loud or background noises, early reflux and feeding difficulties with textural sensitivity, poor eye contact and socialization with aggressive behaviors in preschool. Gestational, medical, and family histories were otherwise normal and physical examination showed no facial or physical anomalies with growth at the 50th centile for height and head size, 80th centile for weight.
Commercial microarray analysis was ordered by the physician and showed missing material at band 11.2 of the short (p) arm of chromosome 16—microdeletion 16p11.2, described in the report as arr cgh chr16:28,503,603-30,283,067x1; arr cgh indicates the technique of array-comparative genomic hybridization or microarray analysis, the span of the missing DNA segment (dosage x1 rather than normal x2) in base pair coordinates (28,503,603 to 30,283,067) for chromosome 16 as defined by the human genome project. Current WES technology determines gene/DNA dosage as well as DNA sequence changes although microarray is often ordered as a separate test in children with the birth defect patterns suggestive of chromosome disorders.
Microdeletion 16p11.2 qualifies as a known pathogenic (disease-causing) change, associated with a pattern of developmental delays with autistic behaviors and obesity [8]. It also suggested parental studies to exclude mild expression and risks for future children, a family study ideally coordinated or explained by the primary physician. Equally important would be counsel that an autism diagnosis must be confirmed by usual assessment scales rather than being inferred from a genomic test [9].
A 2-year-old boy had frequent respiratory infections despite normal gestational, birth, and developmental histories. Bruising and a nosebleed had occurred with several of these infections and a fourth infection in three months with petechiae over his upper body and trunk led to pediatric evaluation. Values for hemoglobin of 11 g/dL, white blood cell count of 6500/μL with low neutrophils, and platelet count of 5,000/μL led to the presumptive diagnosis of idiopathic thrombocytopenic purpura (ITP). Improved but not normal platelet counts (~ 80,000/μL) were obtained after immunoglobulin and steroid therapy, but continued episodes led the parents to seek genetic evaluation to define the basis of disease and risks for their next pregnancy.
Although mother’s maternal uncle had similar ITP episodes while young, healthy now in his 40s, I felt their risk was only 3-5% for ITP in future children as would be the case for most common multifactorial disorders (diabetes, schizophrenia, isolated birth defects, etc.) that involve many genetic and environmental factors. The parents proved wiser and asked for genetic testing, WES showing that the WAS gene associated with Wiskott-Aldrich syndrome [10,11] had a DNA letter change at nucleotide 741 that altered its code from polar glutamic acid (glu or E in amino acid code) to non-polar alanine (ala or A) at position 236 of the WAS protein (p.Ala236Glu or p.A236E). The mutation was reported as definitely pathogenic because 1) an identical mutation [10] had been observed in a child with Wiscott-Aldrich syndrome; 2) it was present in his asymptomatic mother and likely her brother with ITP as expected for an X-linked condition; and 3) the boy’s symptoms of frequent infections, thrombocytopenia, and neutropenia correlated well with those of Wiscott-Aldrich syndrome.
The primary physician would be important in coordinating specialty care that included new treatment options [standard bone marrow or novel stem-cell transplant/gene therapy-11] and reinforcing the 25% risk for future children along with reproductive options.
A 28-year-old woman returned to her primary physician after several years of unexplained joint pain and fatigue, discouraged by multiple subspecialty evaluations that found no diagnosis. She began having joint pain as a child, dismissed as “growing pains” by her parents and physicians, and had a long history of constipation and stomach pain that had been attributed to stress. She had frequent sprains, several fractures, knee ligament tears, and lumbar disc herniation that had required various surgeries. She had persisted in exercise and activity despite her pain but became extremely fatigued after her first pregnancy. She tried to return to work but found that she had trouble making it through the week, often collapsing Friday evenings and remaining exhausted all weekend to the detriment of her husband and new child. She developed episodes of tachycardia and anxiety that had rarely occurred during childhood, and disturbed sleep contributed to her fatigue. Rheumatology found no evidence of autoimmune disease, gastroenterology normal endoscopy except for mild inflammation, and cardiology no signs of arrythmia or structural defects.
Her primary had supported her several years of evaluation and recognized that she had an underlying disease, referring her to genetics. I documented hypermobility on examination and suspected a form of Ehlers-Danlos syndrome (EDS), doing whole exome sequencing that documented a mutation in the collagen type V alpha-1 chain (COL5A1) gene, a common cause of EDS [12,13]. Armed with this DNA diagnosis the patient and her primary were able to find new subspecialists who recognized her osteoarthritis due to joint laxity, irritable bowel syndrome (IBS) with constipation/bowel immotility, and postural orthostatic tachycardia syndrome (POTS) causing tachycardia, insomnia, and chronic fatigue [12]. Therapy for these complications from autonomic nervous system imbalance included beta-blockers with fluid-salt and gluten/dairy exclusion dietary strategies that restored the woman’s work and family life [13-20].
- https://www.poets.org/poetsorg/poem/his-coy-mistress
- Teer JK, Mullikin JC (2010). Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 19: R145-R151
- Wyandt HE, Wilson GN, Tonk VS (2017) Chapter 11: Gene and genome sequencing: Interpreting genetic variation at the nucleotide level. In: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, ( 2nd Edn), Springer Nature, Singapore.
- Wilson GN (2014) Presymptomatic and preimplantation genetic diagnosis: Neurology, NextGenetics, and the next generation. JAMA Neurol 71: 403-404.
- www.omim.org
- https://decipher.sanger.ac.uk/
- www. ncbi.nlm.nih.gov/; http://oct2012.archive.ensembl.org/Homo_sapiens/; https://genome.ucsc.edu/
- Shinawi M (2010) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 47: 332-341.
- Wilson GN, Tonk VS (2011) Autism and genetic testing: An update for clinical practice. Consultant Pediatricians 10: 350-356.
- Villa A (1995) X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nature Genet 9: 414-417.
- Boztug K. (2010) Stem-cell gene therapy for the Wiskott-Aldrich syndrome. New Eng J Med 363: 1918-1927.
- www.oatext.com/joint-laxity-hypermobility-old-problems-and-new-opportunities-for-family-medicine.php
- Bloom L (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Mardis ER (2011) A decade’s perspective on DNA sequencing technology. Nature 470:198-202.
- International Human Genome Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409: 860-921.
- Venter JC (2001) The sequence of the human genome. Science 291: 1304-1351.
- Kharrazi M (2016) Evaluation of a new newborn screening model for cystic fibrosis. J Pediatr 175: 7-9.
- Futreal PA (1994) BRCA1 mutation in primary breast and ovarian carcinomas. Science 266: 120-122
- Pryor JL (1997) Microdeletions in the Y chromosome of infertile men. New Eng J Med 336: 534-539
- Norton ME, Jacobsson B, Swamy GK (2015) Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med 372: 1589-1597.